Browse through our Journals...
Alzheimer’s disease: Role of oxidative stress and its treatment with antioxidants
Harikesh Dubey* (a), Anamika Singh (b), Vishal S. Zine (a), Angad M. Patole (a).
(a) Institute of Pharmaceutical Education and Research , Wardha (M.S.), India-442001.
(b) Raj Kumar Goel Institute of Technology, Ghaziabad (U.P.), India- 201001
Abstract
Oxidative stress plays an important role in the pathogenesis of Alzheimer disease (AD), is an age related neurodegenerative disease and characterized by dementia. AD is associated with β-amyloid accumulation deposited extracellularly in the brains of patients with AD, presence of amyloid plaques and intracellular neurofibillary tangles (NFT) causes mild cognitive impairment (MCI). Amyloid β peptide is capable of generating free radicals. Oxidative mechanisms are involved in the cell loss and other neuropathology associated with AD is evidenced by the large number of metabolic signs of oxidative stress as well as by markers of oxidative damage. Oxidative damage causes nucleic acid oxidation, protein oxidation and lipid peroxidation in neuron cells resulting production of their oxidative products [like: 8-hydroxy-2’-deoxyguanosine (8-OHdG), malondialdehyde, peroxynitrite, 4-hydroxynonenal (4-HNE)], used as a markers in diagnosis of AD. Reactive oxygen species (ROS) mainly generated in mitochondria, activating its antioxidant mechanism thus mitochondrial antioxidants [Glutathione (GSH), superoxide dismutase (SOD) and catalase (CAT)] have protective role, are reduced significantly in AD. Antioxidant like vitamin E, estrogens and red wine have a protective effect on oxidative stress induced neuronal degeneration, may be used in the treatment of AD.
Pathology of Alzheimer disease
Alzheimer’s disease is a complex neurodegenerative disease. Neuropathology of AD involves destruction of neurons in the cortex and limbic structures of the brain responsible for higher learning, memory, reasoning, behavior, and emotional control. Anatomically, the cortical atrophy degeneration of cholinergic neurons characterized by progressive accumulation of paired helical filaments (PHF) as NFT in neurons, amyloid fibers in neuritic (senile) plaques and in the walls of blood vessels is four major alterations in brain structure [1].
Amyloid Protein
The major amyloid protein in AD and also in adult Down syndrome (DS) is amyloid β-protein (Aβ) of 39–43 amino acids. Aβ exists in both soluble and fibrillar forms. Soluble Aβ is a normal metabolic product, present in cerebrospinal fluid (CSF), sera of normal individuals and patients with AD. A unique feature of Aβ is its ability to aggregate and to make insoluble fibrils that are deposited extracellularly in the brains of patients with AD [2].
NFT pathology in AD is associated with hyperphosphorylation of tau. Tau is a neuronal microtubule associated protein and is regulated via its phosphorylation by various protein kinases. Normally 2–3 moles of phosphates per mole of tau are present, whereas this protein is three to four fold hyperphosphorylated in AD [3,4]. This abnormally hyperphosphorylated tau becomes incompetent for microtubule assembly and, consequently, aggregates into NFT containing PHFs [3, 5-7]. Although both NFT and amyloid pathology are common features of AD, it is not clear how these two pathologies relate to each other. However, both pathologies are the end result of aggregation of proteins, Aβ gets fibrillized and makes amyloid plaques, while tau gets aggregated upon hyperphosphorylation, resulting in NFT pathology. Extensive evidence suggests that these two pathological hallmarks of AD may have a link with free radical-mediated assault, i.e. oxidative stress.
Various mechanisms of neuronal degeneration in AD have been proposed, which include formation of free radicals, oxidative stress, mitochondrial dysfunction, inflammatory processes, genetic factors, environmental impact factors, apoptosis, and so on. These factors may interact and amplify each other in a vicious cycle of toxicity leading to neuronal dysfunction, cell dysfunction, and finally cell death (Fig-1). Aβ is produced by the proteolytic cleavage of membrane associated β amyloid precursor protein (APP) [8]. Processing of APP occurs by two major protease pathways, first cleavage of APP at the N-terminus of the Aβ region by β−secretase [9], second at the C-terminus by γ− secretase [10, 11] represents the amyloidogenic pathway for processing of APP to form Aβ Alternatively, APP can also be processed by α−secretase [12], which cleaves within the Aβ sequence, and does not produce Aβ. Patients with Down syndrome generally develop AD in middle age because they have three copies of chromosome 21 containing the APP gene.
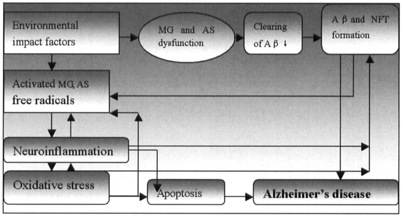
Fig 1 General pathogenesis in Alzheimer’s disease
MG :Microglia ; AS: Astrocyte; AP: Amyloid protein beta; NFT: Neurofibrillary tangles
Alzheimer disease and oxidative stress:
Oxidative stress, in general, is the overpowering of the antioxidative defense system by the oxidative system. Oxidative stress is produced by free radicals, i.e. ROS that are generated by oxygen- and nitrogen-based molecules that have unpaired electrons. Free radicals are very unstable and highly reactive. In order to make paired electrons, free radicals are in constant search of electrons, and therefore, they grab the electrons from other molecules. After loosing the electrons, the donor molecules become unstable and are converted into free radicals themselves. The free radicals produced in the body are toxic, and if not removed or neutralized, they react with lipids, proteins, and nucleic acids in turn damage cellular functions. Generally, oxidative damage to the cellular components results in alteration of the membrane properties such as fluidity, ion transport, enzyme activities, and protein cross-linking. Excessive oxidative damage eventually results in cell death [13].
Experimental models and human brain studies suggest that oxidative stress plays an important role in neuronal degeneration in AD. Soluble Aβ, Aβ fibrils, NFT, mitochondrial abnormalities and aging are contributing factors to increased oxidative stress in AD. Imbalances of oxidative homeostasis leading to increased lipid peroxidation have been revealed as important factors involved in neurodegenerative disorders such as AD. Under normal conditions, ROS are neutralized by the action of the ROS defense system, evidence exists that ROS generation increases with amyloid and NFT pathology in AD, thereby creating a vicious cycle of ROS generation that far exceeds the antioxidant defense system. The brains of patients with AD contain increased levels of lipid peroxidation products such as HNE or 2-propenal (acrolein), and enhanced lipid peroxidation can also be detected in the CSF and plasma of AD patients [14].
ROS-mediated oxidative damage of proteins and nucleic acids is also an important event in AD pathology. There is increasing evidence that both soluble and fibrillar Aβ can induce oxidative stress and ROS are involved in Aβ fibrillization and NFT formation in AD.
Recently high levels of 8-OHdG, a sensitive marker of DNA damage, were found in both nuclear and mitochondrial DNA of the brain in AD suggest that a general imbalance is present in this disease between oxidants and antioxidants [15].
Source of ROS:
Mitochondria are essential organelles for neuronal function. Neurons have the limited glycolytic capacity but energy requirement is high, makes them highly dependent on aerobic oxidative phosphorylation. However, oxidative phosphorylation is a major source of endogenous toxic free radicals, including hydrogen peroxide (H2O2), hydroxyl (·OH), and superoxide (O2·−), that are products of normal cellular respiration [16]. With inhibition of the electron transport chain, electrons accumulate in complex I and coenzyme Q, from which they can be donated directly to molecular oxygen to give O2−· that can be detoxified by the mitochondrial MnSOD to give H2O2 that, in turn, can be converted to H2O by glutathione peroxidase. However, O2·− in the presence of nitric oxide (NO·), formed during the conversion of arginine to citrulline by nitric oxide synthase, can generate peroxynitrite (ONOO−) and lead to protein modification [17]. Furthermore, H2O2 in the presence of reduced transition metals can be converted to toxic ·OH via Fenton and/or Haber Weiss reactions, a process that we have specifically localized to neurofibrillary pathology in AD. Inevitably, if the amount of free radical species overwhelms the capacity of neurons to counteract these harmful species, oxidative stress occurs, followed by mitochondrial dysfunction and neuronal damage. Reactive species generated by mitochondria have several cellular targets, including mitochondrial components themselves (lipids, proteins, and DNA) [18].
Lipid peroxidation in AD:
The reaction of lipids with molecular oxygen is a process known as lipid peroxidation, this involves intermediate oxygen containing free radicals that attack and abstract hydrogen atoms from lipids, particularly polyunsaturated lipids. The majority of lipids are transported in association with lipoproteins in circulation. Oxidation of lipoprotein that carry unsaturated fatty acids, in particular is believed to play a key role in the development and progression of AD [19, 20]. Increased lipid peroxidation is followed by amyloid plaque formation in an animal model of Alzheimer amyloidosis. Extracellular amyloid plaques, intracellular NFT and loss of basal forebrain cholinergic neurons in the brains of AD are the result of abnormalities in lipid metabolism and peroxidation that may be caused, or exacerbated by Aβ [21]. Evidence strongly implicates regionally increased oxidative damage to brain beyond what occurs with aging as one of the processes that may contribute to AD progression.
Apolipoprotein E (ApoE) is the principal apolipoprotein in the central nervous system, and ApoE serves as the major apolipoprotein that is capable of lipid transportation and regulation of lipid metabolism through known receptor-mediated processes. ApoE-dependent dendritic and synaptic regeneration may be less efficient with ApoE4, and this may result in age-related neurodegenerative changes. The increased risk of AD associated with ApoE4 may be modulated by diet, vascular risk factors, and genetic polymorphisms that affect the function of other transporter proteins and enzymes involved in brain lipid homeostasis. Moreover, inheritance of the ApoE4 allele represents the strongest genetic risk factor for sporadic AD. Evidences suggest that ApoE isoforms may specifically influence the cellular distribution of lipid peroxidation products in brain and may therefore contribute to the stratification of risk for AD associated with ApoE4. Furthermore, the 12/15 lipoxygenase (12/15LOX ) enzyme is increased in pathologically affected frontal and temporal regions of AD brains, and the activation of this enzyme occurs early in the course of AD, before the onset of overt dementia, implicating 12/15 LoX mediated lipid peroxidation in the pathogenesis of AD [22].
The lipid peroxidation phenomena could have a major or even causal influence on the pathogenesis of the disease. Some researchers showed that in Down syndrome, which involves a neurodegenerative component similar to or even identical to that of AD, neuronal death occurs according to a process of apoptosis that is related to an increase in lipid peroxidation and can be stopped by catalase and free radical scavengers [23]. A large number of data indicate that a cascade of events contribute to the neurodegeneration in AD.
Metal (Al, Fe, Cu, Zn) dyshomeostasis is believed to play a pivotal role in the pathogenesis of AD [24]. Aluminum (Al3+) has been proposed as one of the critical environmental factors responsible for AD, its ionic or complicated forms that cause oxidative stress in biological systems. Al3+ and aluminum complex also contributes to the neurodegeneration in AD. Al3+ compounds enhance lipid peroxidation in liposomes cellular damage caused by oxidative stress. Al3+ and aluminum complex affected the membrane fluidity on the inner side [25]. Accordingly, multifunctional compounds combining metal chelating and anti-oxidative activity hold a great promise as potential drugs for treating AD [26].
Protein oxidation in AD:
Protein carbonyl content increases in AD hippocampus and in AD inferior parietal lobule relative to AD cerebellum, a brain region showing little degenerative change. There is a significant increase in protein oxidation in frontal pole and occipital pole [27].
Excess brain protein oxidation, as a marker of oxidative stress, contributes to the pool of damaged enzymes in AD. A research reported that elevated LDL in AD was correlated with brain AP-42 levels. Paraoxonase l (PON1) is a calcium-dependent esterase, which contributes to the antioxidant protection conferred by high density lipoprotein on LDL oxidation. Glycation phenomena have been held responsible for many age related pathologies [28, 29].
Glycation of proteins starts as a nonenzymatic process with the spontaneous condensation of ketone or aldehyde groups of a sugar with a free amino acid group to form a labile Schiff base. A cascade of reactions then result in the formation of advanced glycation end products (AGEs ), which are composed of irreversibly cross-linked heterogeneous protein aggregates. AGE receptor, known as RAGE, is also a β-amyloid receptor [30, 31]. The combination of AGEs and RAGE can cause oxidative stress, as shown in the production of thiobarbituric acid reactive substances, heme oxygenase-1, and the activation of nuclear transcription factor kappaB (NF-kB). β Amyloid binding with RAGE also elicits the macrophage colony stimulating factor [32, 33]. This introduces an inflammatory pathway, according to a procedure linking oxidative processes and inflammation in AD [34].This might be a partial explanation for the effect of nonsteroidal antiinflammatory drugs, their NF-kB blocking action [35].
Additionally, several studies have identified within the brains of AD patients, particularly in the NFT, the end products of peroxidation: malondialdehyde [36], peroxynitrite [37], carbonyls [38], AGEs [39], SOD [40, 41] and heme oxygenase [42, 43]. Heme oxygenase-1 is a cellular enzyme that is up-regulated in the brain and in other tissues in response to an oxidative challenge or other noxious stimuli. Protein oxidation has also been observed in elderly individuals with and without AD, but appears to be more marked in AD patients in the regions presenting the most severe histopathologic alteration [44].
Nucleic acid oxidation in AD:
Nucleic acids (nuclear DNA, mitochondrial DNA, and RNA) are one of the several cellular macromolecules damaged by ROS, particularly the hydroxyl radical. Because neurons are irreplaceable and survive as long as the organism does, they need elaborate defense mechanisms to ensure their longevity. In AD, however, an accumulation of nucleic acid oxidation is observed, indicating an increased level of oxidative stress and/or a decreased capacity to repair the nucleic acid damage [18].
DNA oxidation:
DNA is the primary target of ROS, leading to cellular aging. As brain has high oxygen consumption rate, ROS may contribute to neuronal damage in aging and neurological disorders. The hydroxyl radical plays a major role in DNA oxidation, because the copper ion, through the Fenton reaction participates in the attachment of DNA [45]. Formation of modified DNA bases could result in alterations in replication of DNA or inappropriate base pairing producing mutations that could lead to altered protein synthesis. It has been reported that ·OH reacts with purines to form mutagenic 8-hydroxypurine and putatively nonmutagenic formamidopyrimidine lesions. The formamidopyrimidine lesions inhibit DNA synthesis [46, 47]. Because 8-hydroxypurine has the lowest oxidation potential of the four DNA bases, guanine is the most readily oxidized base through free radical attack of C8 leading to the formation of 8OHG under elevated oxygen tension so it is most commonly used markers for DNA oxidation and 2,6- diamino-4-hydroxy-5-formamidopyrimidine (fapyguanine) under conditions of reduced oxygen tension [48,49,50].
In mitochondrial DNA (mtDNA) oxidation, especially 8OHG is of a much greater magnitude than nuclear DNA (nDNA). nDNA gains some protection from histones and morphologically is not close to large oxidant generation. In contrast, mtDNA does not have protective histones, is in close proximity to oxidant generation, may not have a rich antioxidant system, and has relatively limited DNA repair capacity, which may account for some of its vulnerability to oxidation. The accumulation of oxidative modified nDNA and mtDNA results from an imbalance between the rate of oxidation and DNA repair mechanisms.
To counteract the multiple factors causing DNA damage, three DNA repair pathways have evolved. The most prominent repair pathways are base excision repair (BER), nucleotide excision repair, and mismatch repair. BER is characterized by the excision of nucleic acid base residues in the free form that contain lesions including oxidative damage, alkylating adducts, and deamination products. Nucleotide excision repair removes damaged nucleotides as part of fragments up to 30 bases in length. Mismatch repair corrects single misrepaired nucleotides and smaller loops. The inactivation or malfunction of DNA repair mechanisms leads to the accumulation of damaged DNA that characterizes several pathological conditions, including AD [28].
DNA bases are also attacked by the lipid peroxidation products HNE and acrolein, which leads to the formation of bulky exocyclic adducts. This modification can cause inappropriate base pairing that alters protein synthesis. DNA oxidation has been shown to escalate with age, shown by an increase in 8-OHdG in the cerebral cortex and cerebellum brain regions. Similarly both mtDNA and nDNA have increased 8-OHG, 8-hydroxyadenine, and 5-hydroxyuracil in temporal, parietal, and frontal lobes in AD. Overall, mtDNA shows an approximately 10-fold higher intensity of oxidized bases than nDNA. nDNA and mtDNA undergo extensive oxidative damage in AD, which may contribute to the neurodegenerative pathology of this disorder [51].
ROS and hydroxyl radical interaction with DNA generates various products of DNA bases, such as 8-oxo-7, 8 dehydro-2’-deoxyguanosine, 2, 6-diamino-4-hydroxy-5-formamimodiprymidine, 8-OH-adenine, 2-OH-adenine, thymine glycol, and cytosine glycol as shown in fig.2 [1].
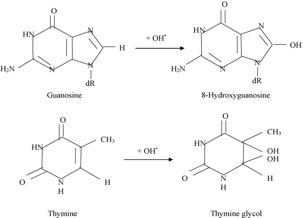
Fig. 2, Formation of the two best-studied oxidation base products of DNA by OH• radical attack. Guanosine: deoxyribose derivative of guanine; dR,, deoxyribose; OH•, hydroxyl radical.
RNA Oxidation:
Oxidative stress damage more easily RNA as compared to DNA, because (unlike DNA) RNA is mostly single-stranded and its bases are not protected by hydrogen bonding. Furthermore, RNA is not covered with protective histones [52, 53]. Oxidative injury to RNA may interfere with correct base pairing, compromise the accuracy of transcription and translation, thus prejudicing normal protein synthesis; it could also promote protein aggregation [53, 54]. Messenger RNA (mRNA) oxidation may be an important factor initiating the cascade of neurodegeneration [55]. RNA species are easily attacked by the hydroxyl radical (.OH), RNA oxidative damage has been performed on the CNS (brain tissue, CSF) and serum of patients with AD. Studies have evaluated 8-hydroxyguanosine (8-OHG) as a marker of hydroxyl radical damage to RNA [52], marked accumulation of 8-OHG from cytoplasmic RNA in the neurons of patients with AD (particularly in the hippocampus, and frontal, temporal and occipital neocortex) have been demonstrated [56]. Neuronal 8-OHG immunoreactivity displayed a significant negative correlation with the duration of the illness and the extent of Aβ deposition, thereby suggesting that RNA oxidative damage preceded pathological changes in AD [57].
Several oxidized mRNA coded for proteins which are believed to be implicated in AD pathogenesis, such as presenilin-1(PS-1), peptides involved in free radicals regulation and detoxification (e.g. Cu/Zn SOD, Carbonyl reductase 1), cell metabolism (e.g. Calpain), and signal transduction (e.g. MAPK kinase 1) [58]. Ribosomal RNA (rRNA) in neurons is more abundant compared to mRNA, and rRNA easily binds to redox-active iron, which promotes rRNA oxidation through the Fenton reaction. Recent study demonstrated that in hippocampal neurons from AD patients, a significant increase in redox active iron which in cytoplasm was mainly associated with rRNA. Furthermore, increased levels of 8-OHG were detected in rRNA [59, 60]. Increased RNA oxidative damage mainly affected rRNA and it was associated, both in AD and MCI, with a significant depletion of rRNA and transfer RNA (tRNA), impairment in ribosome function and decreased protein synthesis [61]. The concentration of 8-OHG in CSF from AD patients was approximately fivefold compared with controls, and it decreased significantly with the duration of the illness and the progression of cognitive dysfunctions, suggesting that RNA oxidation is an early event in AD. RNA oxidation is more prominent in cases with a smaller amount of Aβ plaque deposition or a shorter duration of the disease [62]. RNA oxidation in the AD brain could render the cell incapable of initiating protein synthesis, hindering the cell's defense against further oxidative damage, an effect observed in AD [63].
Biomarkers in AD:
The diagnosis of AD is largely based upon clinical assessment, with definitive diagnosis still requiring pathological evaluation at autopsy. The identification of biomarkers for AD would allow for a less invasive and more accurate diagnosis in the antemortem period. Additionally, biomarkers may facilitate early diagnosis, which is particularly difficult given that there are no signs or symptoms unique to AD. More importantly, they may allow for the identification of individuals with preclinical AD [64].
Peripheral biomarkers:
Significant biological changes related to a condition of oxidative stress have been found not only in brain tissue but also in peripheral tissues of AD individuals. Even there are increasing level of soluble peripheral biomarkers of oxidative stress in biological fluids, mainly CSF but also peripheral blood (PB) (serum/plasma) or urines or even in peripheral tissues themselves such as fibroblasts or blood cells [65]. Elevated levels OF HNE, F2-isoprostane and specific isoprostane 12-iso-iPF2alpha-VI were observed in the CSF, plasma and urines of AD individuals. Studies showed that increase in lipoperoxidation products, malondialdehyde (MDA), and 4- HNE in fibroblasts and lymphoblasts of familial Alzheimer’s disease (FAD) patients. Significantly higher lymphocyte concentration of the oxidized purine 8-OHdG at DNA level, besides a significantly lower plasma levels of antioxidants in AD compared to controls [66].
If altered proteolytic processing of APP underlies AD, then measures of APP or its derivatives may serve as diagnostic markers. Low levels of CSF Aβ42 are an excellent marker for the presence of neocortical amyloid deposition, in the presence or absence of dementia [65]. Aβ42 and tau are specific markers of AD pathogenesis. A recent study has investigated the utility of a marker of neuronal death in the diagnosis of AD, Visinin-like protein 1 (VLP-1), a cytoplasmic calcium sensor protein that is thought to leak from damaged or dying neurons, was found to be significantly increased in the CSF of AD [67]. CSF interleukin-6 (IL-6) has been found to be increased in AD patient as compare to control [65]. Fig: 3 illustrated level of different biomarkers with changes in time at different clinical stages.
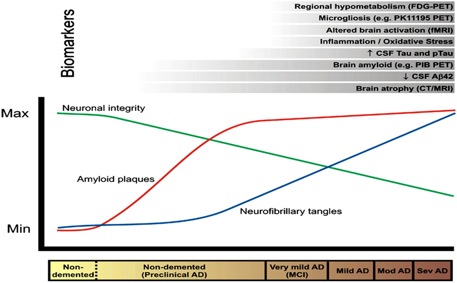
Fig: 3; Hypothesized relationship between the time course of changes in various biomarkers in relation to the neuropathology and clinical changes of Alzheimer's disease.
Proteomics:
Proteomic studies typically include protein preparation by two dimensional gel electrophoresis, liquid chromatography, or protein-chip arrays, followed by mass spectrometry (MS) or tandem MS and database searches to determine protein identity. A newer field of biomarker studies approaches of investigating levels of a single, or several, candidate biomarkers that have been implicated in the pathogenesis of AD, and discover novel biomarkers. As a result of improved MS techniques, proteomics has emerged as a powerful tool for biomarker discovery. Recent efforts to characterize the human CSF proteome have identified 2594 proteins [68], 563 peptide forms and 798 proteins [69], using a combination of approaches. By comparing the differences in protein expression levels between AD and control CSF samples, a number of studies have identified potential diagnostic markers in AD.
Imaging biomarkers
Neuroimaging techniques have potential as markers of disease progression, monitors of therapeutic effects, and predictors of future dementia prior to symptoms in AD. Hippocampus and entorhinal cortex of AD patients, have frequently been targeted by imaging techniques. Atrophy of the medial temporal lobe has been used as a biological marker of AD using MRI with 85% specificity in patients with mild AD. Increased brain atrophy has also been observed in patients at risk for AD who later developed the disease, indicating structural MRI may aid in early diagnosis of the disease. A study found a decrease in hippocampal volume in individuals homozygous for the ε4 allele. These results did not reach significance, however, while changes in glucose metabolism were significant, indicating that changes in brain volume may represent a later marker of disease pathology [70].
Positron emission tomography (PET) has been employed in many AD studies to examine regional cerebral metabolism using 18F-2-deoxy-2-fluoro-D-glucose as a marker (CMRglc using FDG-PET). Observed changes in AD brains include decreased metabolism in temporoparietal, posterior cingulate, hippocampal complex, medial thalamic regions, and mamillary bodies. The first probe for imaging amyloid plaques that was used in living patients was 2-(1-{6-[(2-[F-18] fluoroethyl) (methyl) amino]-2-naphthyl} ethylidene) malononitrile, or [18F]FDDNP [65].
PET imaging study of [18F] FDDNP in subjects with AD, mild cognitive impairment (which may progress to AD), and no cognitive dysfunction, reported significantly higher binding in subjects with AD than those with MCI or control subjects and higher binding in MCI patients than in control subjects, indicating the potential use of this compound in the early diagnosis of AD [71].
The most successful of the imaging amyloid agents has been 11C-labelled Pittsburgh compound B, or PIB, (2-[4′-(methylamino) phenyl]-6-hydrobenzothiazole). In AD, PIB retention is increased in the frontal, parietal, temporal, and occipital cortices and striatum, and studies have consistently shown that nearly all patients diagnosed with Alzheimer's dementia test PIB positive.
Antioxidants and Alzheimer disease
Antioxidant enzymes:
ROS mainly generated in mitochondria that cause oxidative damage to neurons and progression of AD. ROS also may lead to structural and functional abnormalities in mitochondria, also the mitochondrial dysfunction in aged and AD patient. Thus mitochondrial antioxidants play an important role in treatment of AD [72]. An antioxidant defense system protect the brain from oxidative damage, these include antioxidant enzymes and free radical scavengers such as ascorbate, vitamin E, and protein sulfahydryls.
Recent experimental study performed in triple-transgenic mouse, suggest that oxidative stress occurs early in the development of the AD, level of antioxidants, namely, reduced glutathione and vitamin E, are decreased and the extent of lipid peroxidation is increased. Also increased activity of the antioxidant enzymes glutathione peroxidase and SOD before the appearance of Aβ plaques, NFT and cognitive impairments occurs [73].
Impairment in cellular total antioxidant capacity (TAC) plays a central role in AD. Decline in the GSH levels are changed in specific regions of the central nervous system, initiates oxidative stress-mediated neuronal loss of AD patients. Recently, it has been found that a significant reduction in GSH content in lymphoblasts carrying APP, PS-1 and presenilin-2 (PS-2) gene mutations [74]. Glutathione redox status, properly represented by the ratio of oxidizes glutathione and reduced glutathione (GSSG/GSH¬) constitutes a reliable index of oxidative stress, this ratio in red blood cells shows a useful indicator of oxidative stress in several physiological and pathophysiological situations. The GSSG/GSH ratio in Alzheimer’s disease patients, was found significantly increased as compared with their age matched controls [75].
It has been reported that SOD activity is decreased in hippocampus, cerebellum and frontal cortex from AD patients [76]. The activities of the antioxidant enzymes Cu/Zn SOD and CAT are significantly reduced in the frontal and temporal cortex of AD patients [74]. In the hippocampus and inferior parietal lobule, but not in cerebellum of AD patients, the levels of CAT, GSHPx, and GSSG-R mRNAs were elevated, which may reflect the protective gene response to an increased peroxidation in the brain regions showing severe AD pathology. Studies of antioxidant enzymes in AD show controversial results about the increase or decrease in their activities, suggesting that the origin and development of AD is not necessarily due to a failure on the antioxidant enzymatic defenses [76].
SOD-1 activity increased in the cerebrospinal fluid (CSF) with aging has been reported in AD, suggested that oxidative stress increased with time then protective mechanism or apoptosis were induced due to oxidative stress activated response, it is ensure that neurons do not rapidly succumb to oxidative stress [77].
Recent study reveals that aged rat treated with Acetyl-l-carnitine (ALCAR) and R-alpha-lipoic acids (LA) were able to reduce oxidative stress and restore cognitive function and mitochondrial structural abnormalities in all parenchymal cells. One more study demonstrated that significantly decreased endogenous antioxidants and less SOD activity, more oxidative damage to lipids and proteins, and decreased activities of the mitochondrial complex I, IV and V in the brain mitochondria of old rats as compared to young rats [72].
Antioxidant vitamins:
Involvement of oxidative stress in many neurodegenerative diseases, have led to the idea that the therapeutic use of antioxidants could be of help in aging and neurodegenerative diseases. The antioxidant treatments using vitamins, Vitamin E, Vitamin E analogs, and Vitamin C have been well described in literatures. Many vitamins directly scavenge ROS and in parallel can upregulate the antioxidant capacity of oxidative defense system of the body.
Vitamin E:
Among them, Vitamin E or trolox (a water-soluble Vitamin E analog) has been recognized as one of the most important antioxidants also in “in vitro” models such as AD fibroblasts. Vitamin E has been also recognized to inhibit ROS-induced generation of lipid peroxyl radicals, thereby protecting cells from peroxidation [65]. It interacts with cell membranes, traps free radicals, and interrupts the chain reaction that damages cells. It is thought to mitigate the inflammatory effects of plaque formation in the brain. In vitro, Vitamin E protects nerve cells from the effects of Aβ [78]. It decreases the oxidative stress and improves the cognitive function in AD patient. Vitamin E has several other cellular functions apart from its antioxidant function. Therefore, the eventual beneficial effects of vitamin E on AD could be due to other functions related to cell signaling rather than its antioxidant properties [75].
Other antioxidants:
Selegiline:
It is a monoamine oxidase inhibitor, similarly to vitamin E may have beneficial effects in patients with AD. It also increases levels of catecholamines, and adrenergic stimulation may improve cognitive deficits associated with AD. (Various studies have examined evidence for the use of selegiline, in the treatment of AD) [78].
Steroid hormones:
Steroid hormones have antioxidant neuroprotective properties, estrogens being particularly important, more potent inhibitor of iron catalyzed lipid peroxidation in brain tissue than vitamin E. 17-b-estradiol protects neurons from Aβ-mediated cytotoxicity and from oxidative insults attenuating membrane lipid peroxidation and stabilizing neuronal calcium homeostasis. It has been found that estrogen treatment decreases the Aβ level [76].
Red wine:
Recent study reported the protective effect of red wine on oxidative stress and antioxidant enzyme activities in the brain [79].
Conclusion
AD is associated with many etiologies and pathogenic mechanism. Among these free radical induced oxidative stress, are the most promising. Oxidative stress is a cause of progressive aging and developing disease including neurodegeneration. Oxidative stress is one of the earliest events in AD pathogenesis. The complex nature and genesis of oxidative damage and responses in AD occurs by mitochondrial abnormalities that can initiate oxidative stress. There are following supporting evidences like, increased lipid peroxidation, increasd 4-HNE in ventricular fluid, increased protein and nucleic acid oxidation in the AD patient. Some biomarkers like, presence of AGE, malondialdeyde, carbonyls, peroxynitrile, reduced SOD, GSH and catalase in AD patient, also indicate that oxidative stress plays an important role in the pathogenesis of AD.
Aβ fibrillization is a key event in the amyloid plaque formation in AD. Some study also shows that Aβ is also capable to generate free radicals that mediate neurons degeneration and death. Oxidative stress plays as important a role in AD pathology as the literature suggests then regular intake of antioxidants may be beneficial before the sign and symptoms of the disease are appear, because ROS play an important role in early stage of disease pathogenesis. Neurons can adapt to the initial encounter with oxidative stress by increasing their antioxidant defense enzyme, MnSODs, glutathione. Thus preventing the decline in the antioxidant defense system may provide an effective means for intervention in the development of neurodegenerative disease and the aging process.
In the conclusion, this review reveals that mitochondria targeted antioxidants shows beneficial effects on MCI and the combination of antioxidants might be greater potential in the treatment of AD.
REFERENCES
1. Depiro J.T., Talbert R.L., Yees G.C., Matzke G.R., Weels B.G., Posey L.M. (2005), “Pharmacotherapy – A Pathophysiologic approach”, MCGRAW-HILL Medical Publishing Division, 6th edition,1158-1159.
2. Chauhan V., Chauhan A. (2006), “Oxidative stress in Alzheimer’s disease” Pathophysiology 13, 195–208.
3. Grundke-Iqbal I., Iqbal K., Tung Y.C., Quinlan M., Wisniewski H.M., Binder L.I. (1986), “Abnormal phosphorylation of the microtubule. associated protein tau (tau) in Alzheimer cytoskeletal pathology”, Proc. Natl. Acad. Sci. U.S.A. 83, 4913–4917.
4. Kopke E., Tung Y.C., Shaikh S., Alonso A.C., Iqbal K., Grundke I. I. (1993), “ Microtubule-associated protein tau, Abnormal phosphorylation of a non-paired helical filament pool in Alzheimer disease”, J.Biol. Chem. 268, 24374–24384.
5. Alonso A., Zaidi T., Novak M., Grundke I. I. , Iqbal K. (2001), “Hyperphosphorylation induces self-assembly of tau into tangles of paired helical filaments/straight filaments”, Proc. Natl. Acad. Sci. U.S.A. 98, 6923–6928.
6. Lee V.M., Balin B.J., Otvos Jr. L., Trojanowski J.Q. (1991), “A68: a major subunit of paired helical filaments and derivatized forms of normal tau”, Science 251, 675–678.
7. Iqbal K., Grundke I. I., Zaidi T., Merz P.A., Wen G.Y., Shaikh S.S., Wisniewski H.M., Alafuzoff I., Winblad B. (1986), “Defective brain microtubule assembly in Alzheimer’s disease”, Lancet 2 , 421–426.
8. Kang J., Lemaire H.G., Unterbeck A., Salbaum J.M., Masters C.L., Grzeschik K.H., Multhaup G., Beyreuther K., Muller-Hill B. (1987), “The precursor of Alzheimer’s disease amyloid A4 protein resembles a cell-surface receptor”, Nature 325, 733–736.
9. Vassar R., Bennett B.D., Babu-Khan S., Kahn S., Mendiaz E.A., Denis P., Teplow D.B., Ross S., Amarante P., Loeloff R., Luo Y., Fisher S., Fuller J., Edenson S., Lile J., Jarosinski M.A., Biere A.L., Curran E., Burgess T., Louis J.C., Collins F., Treanor J., Rogers G., Citron M. (1999), “Beta-secretase cleavage of Alzheimer’s amyloid precursor protein by the transmembrane aspartic protease BACE”, Science, 286, 735–741.
10. Sastre M., Steiner H., Fuchs K., Capell A., Multhaup G., Condron M.M., Teplow D.B., Haass C. (2001), “Presenilin-dependent gammasecretase processing of beta-amyloid precursor protein at a site corresponding to the S3 cleavage of Notch”, EMBO Rep. 2, 835–841.
11. Yu C., Kim S.H., Ikeuchi T., Xu H., Gasparini L., Wang R., Sisodia S.S. (2001), “Characterization of a presenilin-mediated amyloid precursor protein carboxyl-terminal fragment gamma. Evidence for distinct mechanisms involved in gamma-secretase processing of the APP and Notch1 transmembrane domains”, J. Biol. Chem. 276, 43756–43760.
12. Vardy E.R., Catto A.J., Hooper N.M. (2005), “Proteolytic mechanisms in amyloid-beta metabolism: therapeutic implications for Alzheimer’s disease”, TrendsbMol. Med. 11, 464–472.
13. Bandopadhyay U., Dipak D., Banerjee R.K. (1999), “Reactive oxygen species: oxidative damage and pathogenesis”, Curr. Sci. 77, 658–666.
14. Arlt S., Beisiegel U., Kontush A. (2002), “Lipid peroxidation in neurodegeneration: new insights into Alzheimer’s disease”, Curr. Opin. Lipidol. 13, 289–294.
15. Mecocci P., Cherubini A., Polidori M.C., Cecchetti R. (1998), “Oxidative stress and lymphocyte in Alzheimer disease”, Arch. Gerontol. Geriatr. suppl. 6, 313-316.
16. Wallace, D. C. (1999), “Mitochondrial diseases in man and mouse”. Science, 283, 1482–1488.
17. Smith M. A., Taneda, S., Richey, P. L.; Miyata, S.; Yan, S. D.; Stern, D. Sayre, L. M.; Monnier, V. M.; Perry, G. (1994), “Advanced Maillard reaction end products are associated with Alzheimer disease pathology”. Proc. Natl.Acad. Sci. USA 91, 5710–5714;
18. Moreira P. I., Nunomura A., Nakamura M., Takeda A., Shenk J. C., Aliev G. (2008), “Nucleic acid oxidation in Alzheimer disease”, Free Radical Biology & Medicine 44, 1493–1505.
19. Mielke M.M., Lyketsos C.G. (2006), “Lipids and the pathogenesis ofAlzheimer’s disease: is there a link ? ”. Int Rev Psychiatry, 18:2 ,173.
20. Lauderback C.M., Kanski J.I., Hackett J.M. (2002), “Apolipoprotein E modulates Alzheimer’s A beta (1-42)-induced oxidative damage to synaptosomes in an allele-specific manner”. Bruin Kes, 924:1, 90.
21. Lane R.M., Farlow M.R. (2005), “Lipid homeostasis and apolipoprotein E in the development and progression of Alzheimer’s disease”. J Lipid Res., 46:5, 949.
22. Yao Y., Clark C.M., Trojanowski J.Q. (2005), “Elevation of 12/15 lipoxygenase products in AD and mild cognitive impairment”. Ann Neui-ol. 58:4, 623.
23. Busciglio J., Yankner B.A. (1995), “Apoptosis and increased generation of reactive oxygen species in Down’s syndrome neurons in vitro”. Nature, 378(6559), 776.
24. Molina-Holgado F., Hider R.C., Gaeta A. (2007), “Metals ions and neurodegeneration”. Biometals, 20:3-4, 639.
25. Kaneko N., Sugioka T., Sakurai H. (2007), “Aluminum compounds enhance lipid peroxidation in liposomes: Insight into cellular damage caused by oxidative stress”. J Znorg Biochem, 101: 6, 967.
26. Zheng H, Blat D, Fridkin M. (2006), “Novel neuroprotective neurotrophicNNAP analogs targeting metal toxicity and oxidative stress : potential candidates for the control of neurodegene’rative diseases [J]. J Neural Transm Suppl,71, 163.
27. Markesbery W.R.(1997), “Oxidative stress hypothesis in Alzheimer’s disase”, Free Radical Biology & Medicine, 23:1, 134–147.
28. Zheng H., Blat D., Fridkin M. (2006), “Novel neuroprotective neurotrophic NAP analogs targeting metal toxicity and oxidative stress : potential candidates for the control of neurodegene’rative diseases”. J Neural Transm Suppl, 71, 163.
29. Arlt S., Kontush A., Muller-Thomsen T. (2001), “Lipid peroxidation as a common pathomechanism in coronary heart disease and Alzheimer disease”. 2 Gerontol Geriatr, 34:6 461.
30 Sato T., Shimogaito N., Wu X. (2006), “Toxic advanced glycation end products (TAGE) theory in Alzheimer’s disease”. Am J Alzheimers Dis Other Demen, 21:3, 197.
31. Geroldi D., Falcone C., Emanuele E. (2006), Soluble receptor for advanced glycation end products: from disease marker to potential therapeutic target”. Curr Med Chem, 13:17, 1971.
32. Huang Y., Liu F., Grundke-Iqbal I. (2005), “NF-kappaB precursor, p105, and NF-kappaB inhibitor, IkappaB gamma, are both elevated in Alzheimer disease brain”. Neurosci Lett, 373:8, 115.
33. Kitamura Y., Shimohama S., Ota T. (1997), “Alteration of transcription factors NF-kappaB and STAT1 in Alzheimer’s disease brains”. Neurosci Lett, 237:1, 17.
34. Gao F., Bales K.R., Dodel R.C. (2002), “NF-kappaB mediates IL-1 beta-induced synthesis/release of alpha2-macroglobulin in a human glial cell line [J]. Brain Res Mol Brain Res,105:1-2, 108.
35. Chong Z.Z., Li F., Maiese K. (2005), “Erythropoietin requires NF-kappaB and its nuclear translocation to prevent early and late apoptotic neuronal injury during beta-amyloid toxicity”. Curr Neurovasc Res, 2:5, 387.
36. Yan S.D., Chen X., Schmidt A.M. (1994), “Glycated tau protein in Alzheimer disease: a mechanism for induction of oxidant stress”. Proc Natl Acud Sci USA, 91:16, 7787.
37. Zhang YJ. Xu YF, Liu YH. (2006), “Peroxynitrite induces Alzheimer-like tau modifications and accumulation in rat brain and its underlying mechanisms. FASEB J, 20:9 ,1431.
38. Sultana R., Perluigi M., Butterfield D.A. (2006), “Protein oxidation and lipid peroxidation in brain of subjects with Alzheimer’s disease : insights into mechanism of neurodegeneration from redox proteomics”. Antioxid Redox Signal, 8:11-12, 2021.
39. Yan S.D., Chen X., Fu J. (1996), “RAGE and amyloid-beta peptide neurotoxicity in Alzheimer’s disease”. Nature, 382:6593, 685.
40. Bayer T.A., Schafer S., Breyhan H. (2006), “A vicious circle: role of oxidative stress, intraneuronal Abeta and Cu in Alzheimer’s disease”. Clin Neuropathol,; 25:4, 163.
41. Maurya O.P., Mohanty L., Bhaduri G. (2006), “Role of anti-oxidant enzymes superoxide dismutase and catalase in the development of cataract: study of serum levels in patients with senile and diabetic cataracts”. J Indian Med Assoc,104:7, 394.
42. Schipper H.M, Bennett D.A, Liberman A. (2006), “Glial heme oxygenase-l expression in Alzheimer disease and mild cognitive impairment”. Neurobiol Aging, 27:2, 252.
43. Song W., Su H., Song S. (2006), “Over-expression of heme oxygenase-l promotes oxidative mitochondria1 damage in rat astroglia”. J Cell Physiol, 206 :3, 655.
44. Palmer A.M., Burns M.A. (1994), “Selective increase in lipid peroxidation in the inferior temporal cortex in Alzheimer’s disease”. Brain Res, 645 :1-2, 338.
45. Collins, A. R. (1999), “Oxidative DNA damage, antioxidants, and cancer”, Bioessays, 21, 238–246.
46. O'Connor, T. R., Boiteux, S. (1988), Laval, J., “Ring-opened 7-methylguanine residues in DNA are a block to in vitro DNA synthesis”, Nucleic Acids Res, 16, 5879–5894.
47. Malins D.C., Hellstrom K. E. (2002), Anderson K. M., Johnson P. M., Vinson M. A., “Antioxidant-induced changes in oxidized DNA”. Proc. Natl. Acad. Sci. USA, 99, 5937–5941.
48. Markesbery W. R., Lovell M. A. (2006), “DNA oxidation in Alzheimer's disease”, Antioxid. Redox Signal 8, 2039–2045.
49. Steenken, S. (1989) , “Purine bases, nucleosides, and nucleotides: aqueous solution redox chemistry and transformation reactions of their radical cations and e- and OH adducts”, Chem. Rev., 89, 503–520.
50. Breen, A. P.; Murphy, J. A. (1995), “Reactions of oxyl radicals with DNA”, Free Radic. Biol. Med., 18, 1033–1077.
51. Butterfield D. A., Reed T., Newman S. F., Sultana R. (2007), “Roles of amyloid β-peptide-associated oxidative stress and brain protein modifications in the pathogenesis of Alzheimer's disease and mild cognitive impairment”, Free Radical Biology & Medicine, 43, 658–677.
52. Fiala E.S., Conaway C.C., Mathis J.E. (1989), “Oxidative DNA and RNA damage in the livers of Sprague-Dawley rats treated with the hepatocarcinogen 2-nitropropane”, Cancer Res, 20 5518–5522.
53. Bregeon, D., Sarasin A. (2005), “Hypothetical role of RNA damage avoidance in preventing human disease”, Mutat. Res. 1–2, 293–302.
54. Szymanski M., Barciszewska M.Z., Erdmann V.A., Barciszewski J. (2005), “Anewfrontier for molecular medicine: noncoding RNAs”, Biochim. Biophys. Acta., 1, 65–75.
55. Shan X., Chang, Y., Lin, C.L. (2007), “Messenger RNA oxidation is an early event preceding cell death and causes reduced protein expression”, FASEB J. 11, 2753–2764.
56. Nunomura A., Perry G., Pappolla M.A., Wade R., Hirai K., Chiba S., Smith M.A. (1999), “RNA oxidation is a prominent feature of vulnerable neurons in Alzheimer’s disease”, J. Neurosci., 6, 1959–1964.
57. Nunomura A., Perry G., Aliev G., Hirai K., Takeda A., Balraj E.K., Jones P.K., Ghanbari H., Wataya T., Shimohama S., Chiba S., Atwood C.S., Petersen R.B., Smith M.A. (2001), “ative damage is the earliest event in Alzheimer disease”, J. Neuropathol. Exp. Neurol., 8, 759–767
58. Shan X., Tashiro H., Lin C.L. (2003), “The identification and characterization of oxidized RNAs in Alzheimer’s disease. J. Neurosci. 12, 4913–4921.
59. Honda K., Smith M.A., Zhu X., Baus D., Merrick W.C., Tartakoff A.M., Hattier T., Harris P.L., Siedlak S.L., Fujioka H., Liu Q., Moreira P.I., Miller F.P., Nunomura A., Shimohama S., Perry G. (2005), “Ribosomal RNA in Alzheimer disease is oxidized by bound redox-active iron”, J. Biol. Chem. 22, 20978–20986.
60. Ding, Q., Markesbery, W.R., Cecarini, V., Keller, J.N. (2006), “Decreased RNA, and increased RNA oxidation, in ribosomes from early Alzheimer’s disease”, Neurochem. Res. 5, 705–710.
61. Lovell, M.A., Markesbery, W.R. (2008), “Oxidatively modified RNA in mild cognitive impairment”, Neurobiol. Dis. 2, 169–175.
62. Abe T., Tohg, H., Isobe C., Murata T., Sato C. (2002), “Remarkable increase in the concentration of 8-hydroxyguanosine in cerebrospinal fluid from patients with Alzheimer’s disease”, J. Neurosci. Res. 3, 447–450.
63. Butterfield D. A. , Reed T., Newman S. F., Sultana R. (2007), “Roles of amyloid β-peptide-associated oxidative stress and brain protein modifications in the pathogenesis of Alzheimer's disease and mild cognitive impairment”, Free Radical Biology & Medicine 43, 658–677.
64. Markesbery, W. William R., Schmitt F., Kryscio R., Davis D., Smith C., Wekstein D. (2006), “Neuropathologic substrate of Mild Cognitive Impairment”, Arch. Neurol. 63, 38–46.
65. Craig-Schapiro R., Fagan A., Holtzman D., “Biomarkers of Alzheimer's disease” Neurobiology of Disease 35 (2009) 128–140.
66. Migliorea L., Fontanaa I., Colognatoa R., Coppedea F., Sicilianob G., Murrib L. (2005), “Searching for the role and the most suitable biomarkers of oxidative stress in Alzheimer’s disease and in other neurodegenerative diseases”, Neurobiology of Aging 26, 587–595.
67. Lee, J., Blennow K., Andreasen N., Laterza O., Modur V., Olander J., Gao F., Ohlendorf M., Ladenson H. (2008), “The brain injury biomarker VLP-1 is increased in the cerebrospinal fluid of Alzheimer disease patients”, Clin. Chem. 54, 1617–1623
68. Pan S., Zhu D., Quinn J., Peskind E., Montine T., Lin B., Goodlett D., Taylor G., Eng J., Zhang J. (2007), “A combined dataset of human cerebrospinal fluid proteins identified by multi-dimensional chromatography and tandem mass spectrometry”, Proteomics 7,469–473.
69. Zougman, A., Pilch B., Podtelejnikov A., Kiehntopf M., Schnabel C., Kumar C., Mann M. (2008), “Integrated analysis of the cerebrospinal fluid peptidome and proteome”. J. Proteome Res. 7, 386–399.
70. Nichols L., Pike V. W., Cai L., Innis R. B. (2006), “Imaging and In Vivo Quantitation of b-Amyloid: An Exemplary Biomarker for Alzheimer’s Disease”, Biol.Psychiatry, 59, 940–947.
71. Small G.W., Kepe V., Huang S.C., Wu H.M., Ercoli L., Siddarth P. (2004), “Plaques and tangle imaging using [F18]FDDNP-PET differentiates Alzheimer’s disease, mild cognitive impairment, and older controls”, Neurobiol Aging 25, S58.
72. Alieva G., Palacios H., Walrafena B., Lipsitt A. E., Obrenoviche M.E. (2009), Morales L., “Brain mitochondria as a primary target in the development of treatment strategies for Alzheimer disease”, The International Journal of Biochemistry & Cell Biology 41, 1989–2004.
73. Resende R, Moreira P. I. , Proença T., Deshpande A., Busciglio J., Pereira C., Oliveira C. R. (2008), “Brain oxidative stress in a triple-transgenic mouse model of Alzheimer disease”, Free Radical Biology & Medicine 44, 2051–2057.
74. Cecchi C., Fiorillo C., Sorbi S., Latorraca S., Nachias B., Bangoli S., Nassi P., Liguri G. (2002), “Oxidsative stress reduced antioxidants defences in peripheral cells from familial Alzheimer’s patients”, Free Radical Biology & Medicine, 33:10,1372–1379.
75. Vina J., Lloret A., Orti R., Alonso D. (2004), “Molecular bases of the treatment of Alzheimer’s disease with antioxidants: prevention of oxidative stress”, Molecular Aspects of Medicine 25 117–123,
76. Miranda S., Opazo C., Larrondo L.F., Francisco J., Ruiz F., Leighton F., Inestrosa C. N. (2000), “The role of oxidative stress in the toxicity induced by amyloid β-peptide in Alzheimer's disease”, Progress in Neurobiology 62, 633-648.
77. Zana M., Janka Z., Kalman J. (2007), “Oxidative stress: A bridge between Down’s syndrome and Alzheimer’s disease”, Neurobiology of Aging 28, 648–676.
78. Silvestrelli G., Lanari A., Parnetti L., Tomassoni D., Amenta F. (2006), “Treatment of Alzheimer’s disease: From pharmacology to a better understanding of disease pathophysiology”, Mechanisms of Ageing and Development 127, 148–157.
79. Montillaa P., Espejob I., Munoza M. C., Bujalancea I., Munoz-Castanedac J. R., Tuneza I. (2006), “Protective effect of red wine on oxidative stress and antioxidant enzyme activities in the brain and kidney induced by feeding high cholesterol in rats”, Clinical Nutrition 25, 146–153.
Copyright Priory Lodge Education Ltd 2010
First Published February 2010
Click
on these links to visit our Journals:
Psychiatry
On-Line
Dentistry On-Line | Vet
On-Line | Chest Medicine
On-Line
GP
On-Line | Pharmacy
On-Line | Anaesthesia
On-Line | Medicine
On-Line
Family Medical
Practice On-Line
Home • Journals • Search • Rules for Authors • Submit a Paper • Sponsor us
All pages in this site copyright ©Priory Lodge Education Ltd 1994-


