Browse through our Journals...
Chordoid meningioma – an uncommon variant of meningioma: A case report.
Dr. Raj Pal Singh, Reader, Pathology Dr. Sukant Garg, Senior Resident, Pathology Dr. Harsh Mohan, Professor & Head, Pathology Dr Gauri Joshi, Senior Lecturer, Surgery
Departments of Pathology and Surgery Government Medical College and Hospital Chandigarh, INDIA
Abstract
Objective and Importance: Chordoid meningioma is one of rare histopathological variants of meningioma with a peculiar chordoma-like appearance and association with various hematologic abnormalities.The authors report with a brief review of literature a case of chordoid meningioma in an elderly male patient, highlighting the clinical, neuroradiological and light microscopic features.
Clinical Presentation
A 68-year-old male patient, known case of hypertension for the last ten years, presented with a four month history of severe bifrontal throbbing headache, increased sleep and loss of appetite for 7 days. There was inability to swallow food and incontinence of urine and bowel for 1 day. MRI revealed a large well marginated extra axial space occupying lesion in right frontoparietal area abbuting on the dura . A diagnosis of possible glioma or possible meningioma was made.
Intervention
The patient underwent a right frontoparietal craniotomy for resection of the tumor which was excised in toto.
Conclusion
The patient is asymptomatic during the three year follow-up period after surgery. CT study performed 6 months after surgery showed complete resolution of the edema without any residual mass lesion.
Key Words
chordoid meningioma, hypertension, frontoparietal
Introduction
Chordoid meningioma is one of rare histopathological variants of meningioma with a peculiar chordoma-like appearance and association with various hematologic abnormalities. A literature survey till 2006 revealed that apart from a few series [1-3], the majority of the documented cases are individual case reports [4-8]. In this case report, we describe a case of chordoid meningioma, highlighting the clinical, neuroradiological and light microscopic features.
Case Report
A 68-year-old male patient, known case of hypertension for the last ten years, presented with a four month history of severe bifrontal throbbing headache. There was a history of increased sleep and loss of appetite for 7 days. There was inability to swallow food and incontinence of urine and bowel for 1 day. Physical and neurological examination was unremarkable. Biochemical and hematological parameters were normal except for an elevated erythrocyte sedimentation rate (15 mm first hour). Axial computed tomography (CT) of the brain revealed a 6x 6 x 0.5 cm isodense lesion with mild perilesional edema in the right temporal region. There was a midline shift towards left side. Compression and obliteration of right lateral and third ventricle was seen. The patient was advised to undergo MRI of the brain to evaluate the exact nature of the lesion. MRI revealed a large well marginated extra axial space occupying lesion in right frontoparietal area abbuting on the dura (Figure 1).
Fig 1. MRI showing a large well marginated extra axial space occupying lesion in right frontoparietal area.
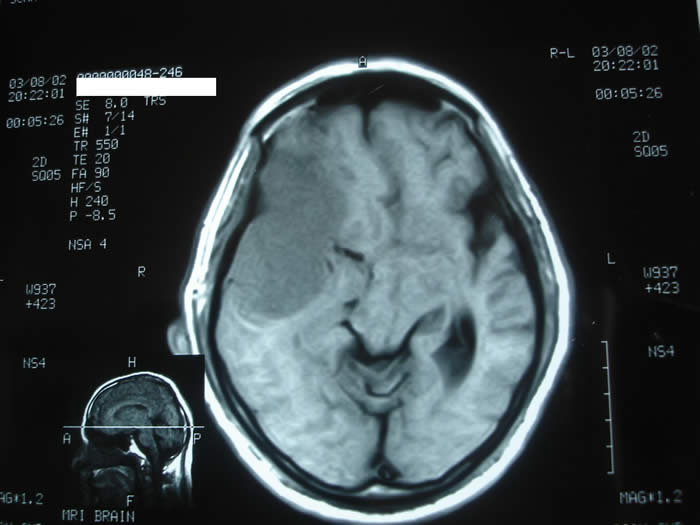
Perilesional edema was present. Ipsilateral ventricular compression and shift to left side was also seen. A diagnosis of ? glioma and ?? meningioma was made and patient underwent a right frontoparietal craniotomy for resection of the tumor. The lesion was seen as a firm globular mass which was adherent to the dura just lateral to the superior sagittal sinus. There was a well-defined gliotic plane around the tumor and the tumor could be easily separated from the brain surface. The tumor was excised in toto and sent for histopathological examination. The defect was closed with temporalis fascia. The entire specimen obtained at surgery was subjected to light microscopic and immunohistochemical studies. Sections revealed sheets, trabeculae and lobules of tumour cells arising from meninges and scattered in a pale basophilic myxoid matrix (Figure 2).
Fig 2. Photomicrograph showing sheets, trabeculae and lobules of tumour cells arising from meninges and scattered in a pale basophilic myxoid matrix (H&E, 200X)
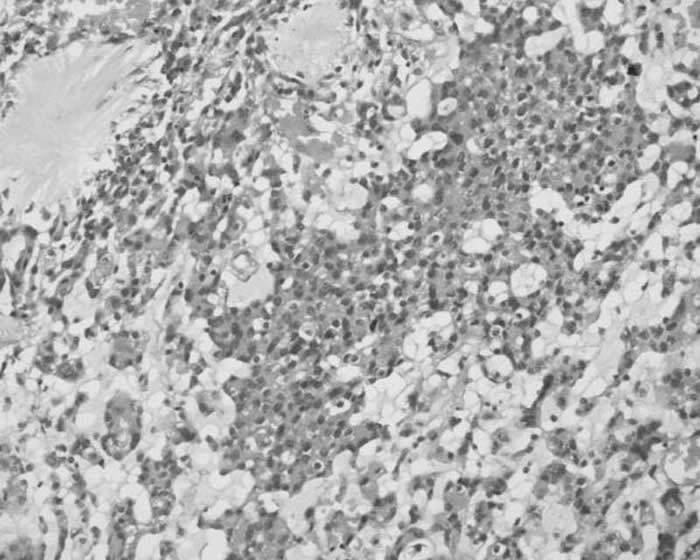
Some of these cells exhibited characteristic cytoplasmic vacuolization. Imperceptibly merging with these epithelial-like cells, were concentric whorl and lobule-like arrangements of cells which were characteristic of meningioma. Occasional psammoma body was also noted. Foci of lymphoplasmacytic infiltration were seen within tumor mass as well as around tumor margin. Neither necrosis nor mitosis was seen. The epithelial-like cells scattered in the basophilic myxoid matrix showed diffuse cytoplasmic staining for vimentin and focal membranous staining for epithelial membrane antigen. These cells showed negative immunostaining for S- 100 and cytokeratin immunostains. Based on the light microscopic and immunohistochemical staining characteristics, a histopathological diagnosis of chordoid variant of meningioma was made.
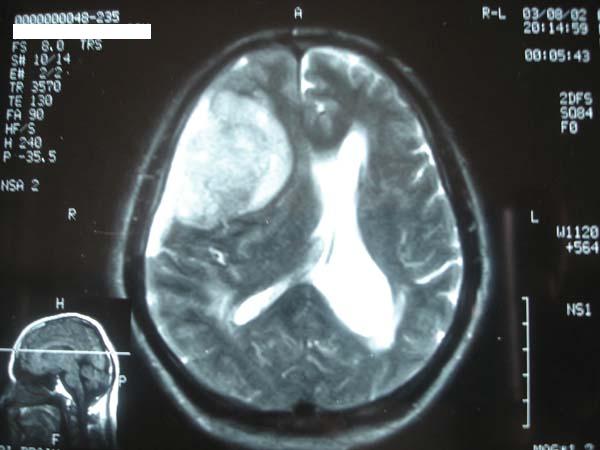
The patient is asymptomatic during the three year follow-up period after surgery. CT study performed 6 months after surgery showed complete resolution of the edema without any residual mass lesion (Fig 3).
Fig 3. CT study showing complete resolution of the edema without any residual mass lesion.
Discussion
Kepes et al [1] for the first time, described a distinct meningeal tumor occurring in children and this was associated with systemic manifestations such as refractory microcytic anemia, hypergammaglobulinemia and angiofollicular lymphoid hyperplasia (Castleman disease). Histopathologically, these tumors were composed of spindle or epithelial cells forming chordoma-like clusters and were scattered in a myxoid matrix. Prominent lymphoplasmacellular infiltration was also a feature in these tumors. Kepes et al [1] also documented in their study that administration of corticosteroids was usually associated with complete resolution of systemic manifestations and a significant decrease in the tumor volume.
In a large series of 42 patients, Couce et al [2] observed that none of their cases had any systemic manifestations as reported by Kepes et al. The mean age of patients in the series of Couce et al was 44 years and hence they expressed the possibility that the systemic manifestations associated with this tumor are limited to chordoid meningiomas occurring in the childhood.
The patient described in this report was an old adult and his main symptom was headache. There were no features suggestive of Castleman disease in our patient. Following a complete surgical excision, the patient became asymptomatic. CT scan performed at 6 months after surgery showed absence of edema and ruled out any residual tumor. The patient was on follow-up for 3 years and has not reported any episodes of headache or seizures. He has been advised to present himself for long-term follow-up.
The precise diagnosis of chordoid meningioma can only be made with the help of an accurate histopathological analysis. Though the histopathological features are distinctive, these tumors should be distinguished from chordoma, myxoid chondrosarcoma, chordoid glioma, metastatic mucinous carcinoma and other variants of meningioma [9-14]. The identification of the classical meningioma component and the demonstration of membrane immunostaining in these tumors will generally establish the diagnosis of chordoid meningioma[8]. However, difficulty in making the diagnosis can still be encountered, particularly for those tumors which arise from the base of the skull. In these cases, differentiation from chordoma may be extremely difficult9. Typical physaliphorous cells, however, are absent in chordoid meningioma. Myxoid chondrosarcoma has not been described in the central nervous system, and is usually positive for S-100 protein [10]. Chordoid glioma is strongly glial fibrillary acidic protein-positive [11]. Microcystic variant of meningioma shows considerable overlap with some features of chordoid meningioma, because it may display certain degrees of myxoid stromal changes and cytoplasmic vacuolation [12,13]. However, this variant lacks inflammatory cell infiltrates. Lymphoplasmacyte- rich meningiomas do not exhibit chordoid features [14].
The recurrence rate in chordoid meningioma is reported to be very high2. In clinical aspect, the chordoid meningioma corresponds to WHO grade II (atypical meningioma) because of its more aggressive clinical behavior, and is recommended to be followed up at regular intervals after surgery [12,15] .Couce et a [2] observed recurrence in all those tumors that were subtotally resected. A possible explanation for the high rate of recurrence could be related to the mucoid quality of its stroma which mechanically
facilitates the spread of the neoplastic cells. This explanation is supported by the fact that all the recurrent tumors in their study were associated with a predominance of a mucin-rich chordoid pattern. Periodic and more close follow-up along with imaging studies after surgery are indicated if resection has been subtotal or if tumor recurrence is suspected on clinical grounds.
References
1. Kepes JJ, Chen WY, Connors MH, Vogel FS. (1988) Chordoid meningeal tumours in young individuals with peritumoral lymphocellular infiltrates causing systemic manifestations of the Castleman syndrome. A report of 7 cases. Cancer, 62, 391-406.
2. Couce ME, Aker FV, Scheithauer BW.(2000) Chordoid meningioma - A clinicopathological study of 42 cases: Am J Surg Pathol , 24, 899-905.
3. Lui PC, Chan TK, Wong SS, Lau PP, Tse GM, Thomas TM, Ng HK. (2000) Cytology of chordoid meningioma: a series of 5 cases with emphasis on differential diagnoses. J Clin Pathol, 12,132-45.
4. Yano H, Shinoda J, Hara A, Shimokawa K, Sakai N.(2000) Chordoid meningioma. Brain Tumour Pathol ,17,153-7.
5. Civit T, Baylac F, Taillandier L, Auque J, Hepner H. Chordoid meningiomas.(1997) Clinical neuroradiological and anatomopathological aspects. Neurochirurgie,43,308-13.
6. Glasier CM, Husain MM, Chadduck W, Boop FA.(1993) Meningiomas in children; MR and histopathologic findings. Am J Neuroradiol ,14,237-41.
7. Civit T, Taillandier, L Baylac F.(1998) Chordoid meningioma. J Neurosurg, 89,686-7.
8 Donato G, Ferraro G, Signorelli F, et al. (2006) Chordoid meningioma: case report and literature review. Ultrastruct Pathol , 30(4),309-14.
9. Mierau GW, Weeks DA(1987). Chondroid chordoma. Ultrastruct Pathol, 11: 731-7.
10. Cybulski GR, Russell EJ, D’Angelo CM, Bailey OT. (1985) Falcine chondrosarcoma:
case report and literature review. Neurosurgery, 16, 412-5.
11. Brat DJ, Scheithauer BW, Staugaitis SM, Cortez SC, Brecher K, Burger PC. (1998) Third ventricular chordoid glioma: a distinct clinicopathologic entity. J Neuropathol Exp Neurol , 57, 28-90.
12. Radner H, Katenkamp D, Reifenberger G, Deckert M, Pietsch T,Wiestler OD.(2001) New developments in the pathology of skull base tumors. Virchows Arch, 438, 321-35.
13. Kobata H, Kondo A, Iwasaki K, Kusaka H, Ito H, Sawada S. (1998) Chordoid meningioma in a child. Case report. J Neurosurg, 88, 319-23.
14. Loiseau H, Pedespan JM, Vital A, Marchal C, Vital C, Cohadon F.(1995)Lymphoplasmacyte-rich meningioma in a child-Case report. J Neurosurg , 83, 1075-9.
15. Kleihues P, Cavenee WK. (2000) Pathology and genetics of tumors of the nervous system (WHO). International Agency for Research on Cancer (IARC Press), Lyon. Am J Surg Pathol, 21, 1455-65.
ADDRESS FOR CORRESPONDENCE:
Dr. Sukant Garg
Senior Resident,
Department of Pathology,
Govt. Medical College, Sector-32A,
CHANDIGARH-160047
© Copyright Priory Lodge Education Limited 2007
First Published June 2007
Click
on these links to visit our Journals:
Psychiatry
On-Line
Dentistry On-Line | Vet
On-Line | Chest Medicine
On-Line
GP
On-Line | Pharmacy
On-Line | Anaesthesia
On-Line | Medicine
On-Line
Family Medical
Practice On-Line
Home • Journals • Search • Rules for Authors • Submit a Paper • Sponsor us
All pages in this site copyright ©Priory Lodge Education Ltd 1994-


