Hereditary Angioedema: A Clinical Review for the General Physician
Jimmy H.C. Gooi, MD, FRCP
Consultant Clinical Immunologist, St James’s University Hospital, Leeds, United Kingdom & Beaumont Hospital, Dublin, Ireland
Address correspondence to:
Department of Immunology
Beaumont Hospital
PO Box 1297, Dublin 9
Ireland
Disclosures: The author has received travel grants from Allergy Therapeutics, United Kingdom; a lecture honorarium from GSK United Kingdom; and an honorarium from ViroPharma Incorporated for authorship of this article.
Introduction
Recurrent angioedema was first described by Quincke (Quincke 1882) and the hereditary form by Osler six years later (Osler 1888). It was only in 1963 that the immunochemical basis of hereditary “angioneurotic” edema was delineated by Donaldson and Evans (1963). Although mental stress is a known precipitating factor, the term angioneurotic is no longer used, and the accepted nomenclature of this group of disorders is hereditary angioedema (HAE).
HAE is now recognized as a heterogeneous group of disorders of the bradykinin-forming cascade and of vascular permeability. The classic and most common forms of HAE, type I and type II, involve mutations of the C1 inhibitor gene (C1INH) (Gosswein et al 2008; Zuraw and Herschbach 2000). More recently, mutations in other genes in the bradykinin pathway have been described involving factor XII (type III), angiotensin-converting enzyme, and aminopeptidase P (Binkley 2010). Angioedema may also be acquired, but this form of recurrent angioedema has been reviewed elsewhere (Zingale et al 2006; Cicardi and Zanichellli 2010) and is not included in this review.
HAE is a rare yet potentially life-threatening inherited disorder with an estimated prevalence of 1 in 50,000 population (Bowen et al 2010). In an online survey of 313 HAE individuals in United States, Germany, United Kingdom, France, and the Netherlands, the average time taken to establish a diagnosis was 8.3 years and required visits to 4.4 specialists. Sixty-five percent of patients were misdiagnosed, and about 20% had unnecessary surgery, including emergency laparotomy. The most common misdiagnosis was allergy in 38% of the survey participants (Lunn et al 2010). Although there may be a selection bias in the participants of the survey, it is evident that there is a lack of awareness of HAE with consequent delayed diagnosis/misdiagnosis and unnecessary treatment and/or surgery. There have been major advances in the understanding and management of HAE over the past few years. This review discusses the clinical syndrome, genetic basis (type I, II, and III), and current management of HAE with a focus on aspects of HAE that may be of particular interest to general physicians and practitioners.
Clinical Syndrome
The hallmark of HAE is recurrent angioedema. The symptoms and signs of HAE are dictated by the site(s) of angioedema. The common sites affected are the skin, gastrointestinal tract, and upper respiratory tract. Cutaneous angioedema and abdominal symptoms occur in 97% of the episodes (Bork et al 2006a). The frequency and severity of these attacks vary considerably, and severity of disease does not appear to correlate with C1-INH levels.
Symptoms usually develop in childhood or adolescence but may manifest in adulthood. Bork and colleagues (2006a) conducted a retrospective analysis of 221 patients with type I and II HAE who experienced 131,110 angioedema episodes. Of the 8443 patient-years described, 5736 were symptomatic. The mean age of symptom onset in this study was 11.2 years (range 1-40 years) (Bork et al 2006a). The C1-INH deficiency was asymptomatic in 5 adults (age 21, 37, 39, 41 and 61 years). Early onset of symptoms and female sex were associated with a more severe course of disease (Bork et al 2006a). The disease severity may differ in affected members of the same family.
Many patients have prodromal symptoms before the onset of angioedema, and these symptoms include fatigue, myalgia, arthralgia, and nonpruritic erythematous rashes such as erythema marginatum (Figure 1) (Agostini et al 2004).
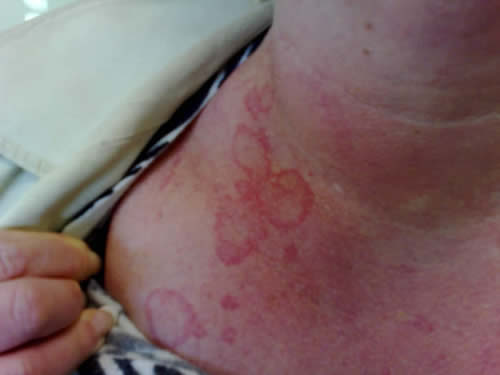
Figure 1. Erythema marginatum, a flat erythematous nonpruritic irregularly shaped rash with central clearing. Erythematous rashes are part of the prodrome, which may develop before or occur concomitantly with angioedema attacks. This patient also shows surrounding erythema of the neck.
Exacerbations may be triggered by infection, trauma, menstruation, stress, oral contraceptives, and ACE inhibitors, but often a triggering factor cannot be identified (Bowen et al 2010).
Cutaneous angioedema involves the deeper dermis and subcutaneous tissues. The swelling is diffuse and ill-defined and not inflamed. The common sites are the extremities, face, and external genitalia but any parts of the anatomy may be affected. Swellings of the face and extremities can be disfiguring and disabling (Figure 2), causing stress and absence from school or work. In tubular visceral organs, the angioedema may be submucosal or transmural.
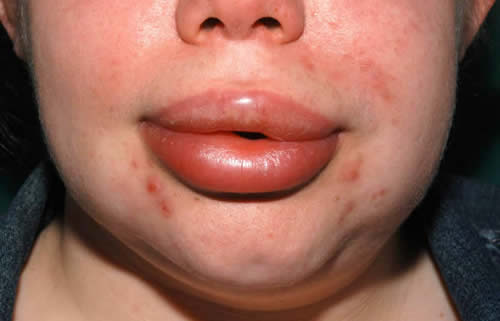
Figure 2. Angioedema (of a few days’ duration) of the lips and lower part of face. Note loss of nasolabial fold.
Free fluid may be detected in serous cavities (Figure 3) and, if severe, detectable as ascites or pleural effusion.
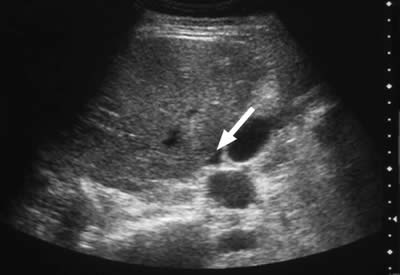
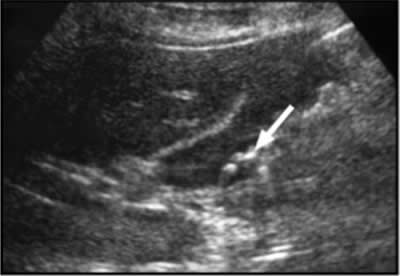
Figure 3. A 30-year-old woman with type I hereditary angioedema (HAE) presented with acute abdominal pain, vomiting, generalised abdominal guarding, and tenderness. Serum amylase was grossly elevated at 2445 IU/L. Abdominal ultrasound indicates free fluid (A) and gallstones (B). The diagnosis of acute pancreatitis and cholelithiasis is confirmed. Free fluid in serous cavities is known to occur in HAE attacks.
Recurrent gastrointestinal angioedema develops in more than 90% of patients (Bork et al 2006a). The initial symptoms may be nonspecific, such as colic, distention, and nausea, eventually leading to vomiting and diarrhea (Bork et al 2006b). Established abdominal angioedema often requires attendance in the casualty department or hospitalization. The presentation can resemble that of an acute abdomen and is often misdiagnosed, leading to unnecessary treatment or surgery (Lunn et al 2010). However, medical and surgical emergencies such as appendicitis (Figure 4), pancreatitis, and cholelithiasis (Figure 3) may occur in HAE patients independently or concomitantly and it is important that such conditions be considered in the differential diagnosis.
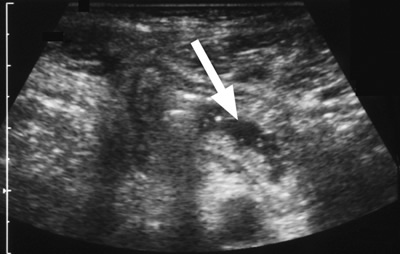
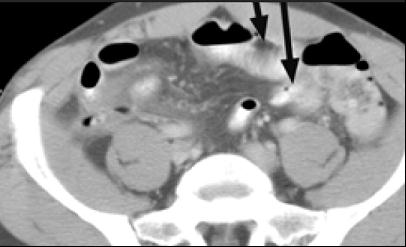
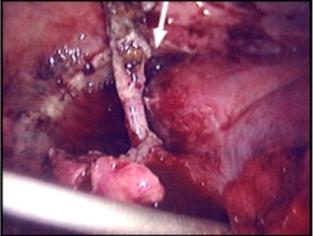
Figure 4. A 48-year-old man with type I HAE presented with abdominal pain, vomiting, fever, and rebound tenderness particularly in the right iliac fossa. Abdominal ultrasound shows mucosal oedema of the small intestine and coin stacking appearance (A) but no free fluid. Abdominal CT shows inflammatory changes in the appendix (long arrow) and caecum (short arrow) (B). Gangrenous appendix is seen at laparotomy (C).
Ultrasound examination of the HAE abdomen often shows mucosal edema and/or a thickened wall of the intestine (Figure 4), and free fluid (Figure 3) or even ascites.
Tightening of the throat, dysphonia, dysphagia, and difficulty swallowing saliva suggest laryngeal angioedema. About one third of cases of facial angioedema extend to laryngeal oedema (Bork et al 2006a). About half of HAE patients will experience a laryngeal attack at some point in their disease course (Bork et al 2006a). If laryngeal angioedema is untreated, the mortality rate from asphyxiation may be as high as 40% (Bork et al 2000a).
Urogenital tract symptoms and headache may develop during an HAE attack but are less common (Bork et al 2006a).
Socioeconomic Aspects
HAE has a significant impact on health-related quality of life, work productivity, and psychological and social well-being. A Web-based survey of 457 patients with HAE in the United States found that 40.5% felt their disease prevented them from advancing in school, and 57.5% felt that the disease affected their career advancement (Lumry et al 2010). HAE patients were also more likely to exhibit depressive symptoms than the general population, with depressive symptomatology increasing with the severity of attacks. In this survey, HAE patients were also more likely to be taking psychotropic medications. HAE also has a significant impact on work productivity, with workers missing a mean 3.3 days of work and students missing an average of 1.9 days of school as a result of their most recent HAE attack (Lumry et al 2010).
Genetics of HAE
The term HAE refers primarily to disorders due to autosomal dominant mutations in the C1-INH gene, located on chromosome 11q11-q13, that lead to a decrease in functional C1-INH. Two immunochemical types of hereditary C1-INH deficiency have been described (Table 1). Type I, which accounts for 80% to 85% of the cases (Nzeako et al 2001), is characterized by a decrease in the level of C1-INH. Type II HAE, comprising about 15% of HAE cases, is characterized by normal or elevated levels of a functionally impaired C1-INH protein that cannot form complexes with target proteases (Gompels et al 2005). There are no known genotype-phenotype correlations. Symptoms are identical in these 2 types of HAE, as is the treatment approach. In about 25% of cases there is no family history of HAE and these cases are thought to be a result of de novo spontaneous mutations (Bowen et al 2010).
Table 1. Types of hereditary angioedema (HAE).
HAE Type |
Genetic Cause |
% of HAE Cases |
Sex Predominance |
C1-INH Level |
C1-INH Function |
C4 Level |
Type I |
Absolute deficiency of C1-INH protein |
~80-85% |
None |
Low |
Low |
Low |
Type II |
Production of a functionally impaired C1-INH protein |
~15%-20% |
None |
Normal or elevated |
Low |
Low |
Type III |
Mutation of factor XII gene or genetic cause other than C1-INH deficiency/loss of function |
~1% |
Women >> men |
Normal |
Normal |
Normal |
A third type of inherited angioedema (type III HAE), first described by Bork et al (2000b), accounts for about 1% of HAE cases and is associated with normal C1-INH level and function (Table 1). Symptoms are similar to those of HAE types I and II (Bork et al 2007). HAE with normal C1-INH function was thought to occur exclusively in women, although it has since been reported to occur in men as well (Martin et al 2007). In some cases, type III HAE results from autosomal dominant mutations in the factor XII gene (this type III HAE variant is designated HAE-FXII), which is located on chromosome 5q33-qter. HAE-FXII appears to be oestrogen-dependent, with oestrogens in oral contraceptives and hormone replacement therapy and pregnancy exacerbating attacks (Bork et al 2010). There are other forms of HAE type III where the genetic basis has not been identified.
Pathogenesis of Angioedema in HAE
The key components in the pathogenesis of oedema in HAE are factor XII (Hageman factor), prekallikrein, kallikrein, high-molecular-weight kininogen (HK), bradykinin, and C1-INH. Factor XII, on binding to an initiating surface, autoactivates to form factor XIIa. In the circulation, prekallikrein is bound noncovalently to HK. Factor XIIa acts on prekallikrein converting it to kallikrein (and also activates factor XI to XIa, involving the intrinsic coagulation pathway). Kallikrein activates factor XII to XIIa and XIIf (which is nonbound) more efficiently than initiating surfaces. Factor XIIf lacks the surface binding region of factor XII and is only capable of activating prekallikrein at sites away from the initiating surface; it also activates the C1r subunit of complement component C1 (Kaplan and Joseph 2010). Kallikrein cleaves HK to form the potent vasoactive nonapeptide bradykinin, which results in increased vascular permeability, vasodilation, and smooth muscle contraction (Figure 5). The evidence suggesting that bradykinin is the mediator of angioedema in HAE includes the presence of kallikrein in induced blister fluids in HAE patients, decreased prekallikrein and HK levels during HAE exacerbations, and increased local and circulating bradykinin levels in acute HAE (Kaplan and Joseph 2010).
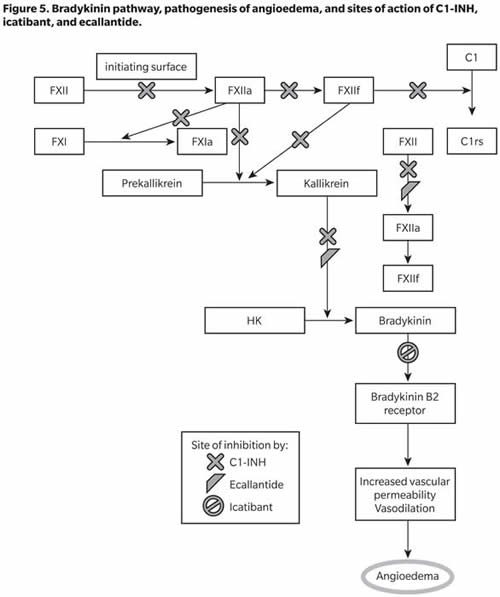
C1-INH inhibits the autoactivation of factor XII and formation of XIIf from XIIa, conversion of prekallikrein to kallikrein, cleavage of HK and further activation of fXII by kallikrein, and activation of C4 and C2 by activated C1. C1-INH, therefore, acts on multiple steps in the bradykinin-forming cascade, and its deficiency leads to increased bradykinin formation and the resulting increased vascular permeability and angioedema. Increased understanding of this cascade has led to the development of several drugs to treat HAE, including C1-INH concentrates (as replacement therapy), icatibant (a bradykinin B2 receptor antagonist), and ecallantide (a kallikrein inhibitor) (Figure 5).
Diagnosis of HAE
HAE should be confirmed or excluded in individuals who suffer from recurrent angioedema (without pruritic rashes or urticaria), chronic recurrent abdominal pain, laryngeal swelling, and/or family history of angioedema. The investigation should start with serum C1-INH and C4 measurement (Figure 6).
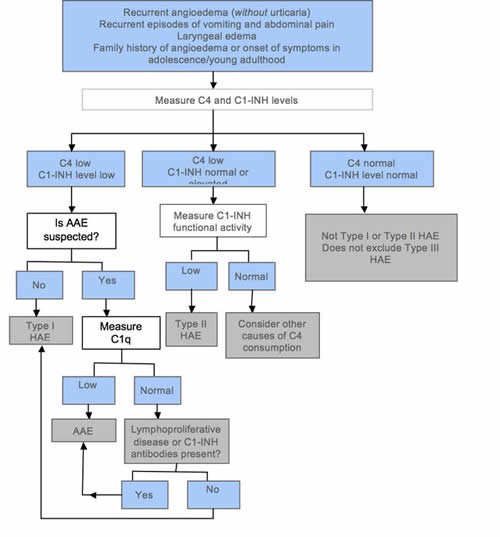
Figure 6. Simplified diagnostic algorithm for hereditary angioedema (HAE). Adapted from Bowen et al (2010).
If both levels are decreased, the results are compatible with type I HAE, provided acquired angioedema (AAE) is not suspected. If AAE needs to be excluded, serum C1q and serum electrophoresis should be requested. If C1q is decreased, the diagnosis is compatible with AAE. C1q is decreased in 70% of AAE cases but is normal in HAE (Cicardi et al 2010). The presence of a paraprotein suggests AAE associated with a lymphoproliferative disease. A normal or slightly elevated C1-INH does not exclude HAE. In such a situation, the functional level of C1-INH should be determined. As this is an assay of C1-INH enzyme function, the sample needs careful handling to minimize the introduction of laboratory artefact. A decreased C1-INH functional level with normal or increased C1-INH immunochemical level and low C4 is compatible with type II HAE. If C4 and C1-INH level and function are normal, HAE types I and II can be excluded, but type III HAE remains a possibility. C4 is almost always decreased in type I and II HAE attacks but may be normal in 2% of asymptomatic patients (Bowen et al 2008). To avoid problems with assay variability, it is prudent to repeat functional C1-INH tests several months later or during an acute episode, particularly if C4 is normal but there is still clinical suspicion of HAE type I or II. It is important to repeat tests when abnormal results are obtained in young children, especially infants. The 2010 International consensus algorithm for the diagnosis, therapy and management of hereditary angioedema does not recommend genetic testing to confirm the diagnosis of HAE (Bowen et al 2010). There are at present no well-validated and robust laboratory test(s) for the diagnosis of type III HAE. The diagnosis is a process of excluding types I and II HAE, drug-associated angioedema, and idiopathic angioedema.
Treatment of HAE and Long-Term Care
Once the diagnosis of HAE is made and confirmed, the patient should be counseled and a care plan developed. The patient should be informed of the medications available for acute and prophylactic treatment of HAE. Informed discussion of situations and drugs that may precipitate an exacerbation of angioedema will be beneficial. The individual should be provided with literature about HAE and patient support/advocacy organizations and informed of the importance of regular follow-ups, which should occur at least twice a year, and as and when necessary. The specialist will often provide a letter for the patient with relevant information about his/her condition, treatment, and contact details in case of emergencies. It is useful to arrange with the patient’s local hospital casualty department a care plan to treat HAE emergencies. The patient is also encouraged to register with an organization like Medic Alert. Pregnancy should be jointly managed with the obstetrics unit. The patient should be made aware of the increased risk of autoimmune disorders (Brickman et al 1986). Because many HAE patients will require therapy with blood or plasma-derived products, baseline laboratory assessments should be performed at diagnosis, including full blood count; liver function tests; virology including hepatitis B and C status; and autoantibodies. (Bowen et al 2010). These tests may be repeated annually and when indicated. If the individual is hepatitis B negative it is advised that he/she be immunized. Hepatitis B antibody status should be checked every 5 years and a booster dose arranged if necessary. The Consensus document recommends a lipid profile and liver ultrasound scan (Bowen et al 2010) for patients who will be treated with attenuated androgens. Abdominal liver and spleen ultrasound should be performed before long-term androgen therapy is started and repeated when indicated.
Treatment of HAE involves 3 components: management of acute attacks, short-term prophylaxis before medical or surgical procedures that are likely to trigger an attack, and routine prophylaxis to prevent HAE attacks in the long term. As discussed, the primary mediator of swelling and other symptoms associated with HAE is bradykinin. Current acute and prophylactic treatments for HAE, therefore, aim to prevent the generation or activity of bradykinin, either by replacing or increasing levels of functional C1-INH; by preventing bradykinin from binding to its target receptor; or directly inhibiting kallkrein, the enzyme responsible for cleaving HK to form bradykinin (Figure 5). Table 2 lists the options available for the acute and prophylactic treatment of HAE.
Table 2. Therapeutic options for acute and prophylactic treatment of HAE attacks.*
Agent |
Description/ Structure |
Licensed Dose |
Most Common Adverse Events |
EMA Licensed Indication |
|
||||
Human C1 esterase inhibitor (Berinert®, CSL Behring, Marburg, Germany) |
Plasma-derived, pasteurized human C1 esterase inhibitor |
20 U/kg body weight by IV injection |
Headache, abdominal pain, nausea, muscle spasm, pain, diarrhea and vomiting |
Approved for acute treatment of HAE; may be self-administered after adequate training |
Human C1 esterase inhibitor (Cinryze®, ViroPharma Incorporated Exton, PA, USA) |
Plasma-derived, pasteurized, nanofiltered human C1 esterase inhibitor |
Acute treatment: 1000 U at first sign of attack onset Preprocedural prevention: 1000 U within 24 hours of a medical, dental, or surgical procedure Routine prevention: 1000 U every 3 or 4 days by IV infusion |
Upper respiratory tract infection, sinusitis, rash, and headache |
Approved for acute treatment and preprocedural and routine prophylaxis in adolescents and adults; may be self-administered after adequate training |
Conestat alfa (Ruconest® or Rhucin®, Pharming NV Leiden, the Netherlands)
|
Recombinant analog of human C1 esterase inhibitor |
50 U/kg body weight by IV injection for adults weighing <84 kg; 4200 U for adults weighing ≥84 kg |
Headache, vertigo |
Approved for acute treatment of HAE in adults |
Icatibant (Firazyr®, Shire UK; Orphan Therapies GmBH Berlin) |
Synthetic non–plasma-derived decapeptide bradykinin B2 receptor antagonist |
30 mg by SC injection Maximum dose 90 mg over 18 hours |
Injection site reactions, rash, nasal congestion, dizziness, headache, asthenia, nausea, abdominal pain |
Approved for acute treatment of HAE in adults; may be self-administered after adequate training |
Ecallantide (Kalbitor®, Dyax Corp Cambridge, MA, USA) |
Recombinant 60-amino acid protein produced in Pichia pastoris yeast cells |
30 mg SC administered in three 10 mg (1 mL) injections. If an attack persists, an additional 30-mg dose may be administered within a 24-hour period |
Headache, nausea, diarrhea, pyrexia, injection site reactions, and nasopharyngitis; hypersensitivity reactions may also occur |
Not approved in EU for acute treatment |
Danazol, stanozolol
|
17-alpha-alkylated attenuated androgens
|
Danazol: Up to 200 mg/day orally Stanozolol: Up to 2 mg/day Titrate to lowest dose |
Weight gain, virilization, muscle pains and cramps, headaches, depression, fatigue, GI side effects (ie, nausea, vomiting, constipation), menstrual irregularities, hepatic side effects |
Unlicensed use of a licensed drug
|
Fresh frozen plasma |
Plasma obtained by apheresis from a carefully selected panel of donors who have been virally screened |
2-3 units (total volume = 400-500 mL) |
Potential transmission of infectious agents. |
Licensed product but no specific licence for acute or prophylactic use |
Treatment of Acute Attacks
Acute attacks of HAE, which can be distressing, disabling, and/or life-threatening, should be treated as soon as possible after symptom onset or reported by the patient (Bowen et al 2010; Cicardi et al 2012). Studies have shown the efficacy of treatment is greater when attacks are treated within 2 hours of onset compared with treatment initiated > 2 hours after attack onset (Bork et al 2005). Specific treatment is advisable for all HAE attacks irrespective of the site affected (Cicardi et al 2012).
Agents available for the acute treatment of HAE include plasma-derived, pasteurized C1-INH concentrate (pdC1-INH); plasma-derived, pasteurized, nanofiltered C1-INH concentrate (nfC1-INH); recombinant human C1-INH concentrate (rhC1-INH); icatibant, a bradykinin B2 receptor antagonist; and ecallantide, a plasma kallikrein inhibitor. Certain specialist centres have programmes that enable patients to self-administer C1-INH concentrate or icatibant outside the hospital setting. Self-administration has the potential to minimize the time between symptom onset and treatment and bring about earlier resolution of symptoms.
Plasma-Derived C1-INH Therapy
A plasma-derived, pasteurized C1-INH concentrate (marketed as Berinert®, CSL Behring) has been used in Germany since 1979 and is now licensed throughout the European Union and is approved for the treatment of all acute HAE attacks at a dose of 20 U/kg body weight by IV infusion. Another plasma-derived C1-INH concentrate available initially in the Netherlands and now in other European countries is Cetor®, manufactured by Sanquin. The recommended dose for this product is 1000 U by IV infusion.
A pasteurized, nanofiltered C1-INH product (marketed as Cinryze®, ViroPharma Incorporated) was recently approved by the European Medicines Agency (EMA) for acute treatment and preprocedural and routine prophylaxis in HAE patients. This product is already approved in the United States for prophylaxis against HAE attacks. The dose for the treatment of acute attacks is 1000 units by IV infusion. A second dose may be administered if the patient has not responded adequately after 60 minutes.
Plasma-derived C1-INH concentrates are effective, well tolerated, and safe, and no viral transmission has been reported to date. These preparations are also safe to use during pregnancy (Bowen et al 2010). The adverse reactions reported include headache, rash, and, rarely, hypersensitivity reactions. Patients can have a supply (1000-2000 U) of C1-INH concentrate at home to self-administer or take to the local casualty department to treat acute HAE episodes.
Recombinant C1-INH Concentrate
Conestat alfa, or recombinant human C1-INH (marketed as Ruconest® or Rhucin® by Pharming) is approved by the EMA for the treatment of acute HAE attacks in adults. The dose is 50 U/kg body weight by IV injection for adults weighing <84 kg. For those weighing ≥84 kg, the dose is 4200 U by IV injection. The adverse reactions reported are headache and vertigo (Zuraw et al 2010). The recombinant C1-INH product is produced from the human C1-INH gene cloned and expressed in mammary glands of transgenic rabbits. Although the risk of hypersensitivity reactions is small, it is important to ensure that patients do not have rabbit allergy before starting therapy (Zuraw et al 2010).
Icatibant
Icatibant (Firazyr®, Shire) is a potent, specific, and selective competitive antagonist of the bradykinin B2 receptor. It is a synthetic non–plasma-derived decapeptide similar in structure to bradykinin but is resistant to enzymatic cleavage (Reshef 2008). Icatibant is approved by the EMA for the treatment of acute HAE attacks in adults and may be self-administered after appropriate training from a healthcare professional. The adult dose is 30 mg, which is administered subcutaneously. Local injection-site reactions are commonly reported. Icatibant may be effective in type III HAE (Bouillet et al 2009), but it is not licensed for the treatment of this condition. Icatibant has not been licensed for use in children and no data on its efficacy and safety in pregnant women have been reported.
Ecallantide
Ecallantide (Kalbitor®, Dyax Corp.) is a recombinant 60-amino acid protein produced in the yeast Pichia pastoris and is a potent inhibitor of plasma kallikrein. It has not been licensed in the European Union but is approved in the United States for the treatment of acute attacks at any location in patients ≥16 years of age. Ecallantide is administered as a subcutaneous injection. Ecallantide is associated with a risk of hypersensitivity reactions, so it must be administered by a healthcare professional. There have been no reports of its use in pregnant women.
Fresh Frozen Plasma
There is little indication for the use of fresh frozen plasma (FFP), given the availability of more disease-specific medications such as C1-INH therapy. FFP was the first effective acute treatment for HAE (Pickering 1969) and was the only definitive therapy until the current generation of medications was developed.
In the unlikely event that C1-I NH concentrate, icatibant, and ecallantide are not available, FFP or solvent/detergent treated FFP can be used effectively for acute and short-term prophylaxis (Prematta et al 2007). The dose is 2 to 3 units (total volume = 400-500 mL), which can be infused rapidly. There have been concerns that FFP treatment may worsen HAE attacks but this was not observed by Prematta et al in their study or in the author’s experience. However, the author has noted transfusion-like reactions rarely when using FFP to treat angioedema attacks.
There are legitimate concerns about the safety of FFP and other blood products. Blood product fractionators currently use plasma obtained by apheresis from a carefully selected panel of donors who have been virally screened. The plasma pool is PCR tested for hepatitis A, B, and C; HIV; and parvovirus B19. Prepared blood products are quarantined for few months before being released for general use. Nevertheless, their viral safety cannot be guaranteed and there are at present no tests for variant Creutzfeldt-Jakob disease.
Prophylactic Treatment
Prophylactic treatment may be required in the short term to prevent HAE attacks during medical/surgical procedures or stressful events or as routine therapy for long-term control of HAE attacks. Medical, dental, and surgical procedures, particularly those that involve manipulation of the upper airways, can trigger potentially life-threatening edema in HAE patients. Prophylaxis may not be required for minor procedures or events that are not likely to induce angioedema, provided an acute treatment is immediately available for use (Bowen et al 2010). However, if acute medication may not be available or if the procedure is likely to induce an attack, prophylaxis with C1-INH therapy is the preferred option (Bowen et al 2010). In the short-term prophylactic setting, studies support the efficacy of doses between 500 and 2000 U for most patients and most procedures (Lumry et al 2011; Rusicke et al 2010). The only C1-INH product that is licensed for preprocedural prophylaxis is nfC1-INH, with a recommended dose of 1000 U administered within 24 hours of a medical, surgical, or dental procedure. Although attenuated androgens, FFP, and antifibrinolytics have been used in patients as short-term preprocedural prophylaxis and are included in the 2010 International consensus algorithm for the diagnosis, therapy, and management of HAE, the current thinking is that these agents have little role in the preprocedural prophylactic setting (Bowen et al 2010; Bowen 2011).
Routine prophylaxis against HAE attacks should be considered for patients who experience more than one severe event per month or if acute treatment is inadequate or not available (Bowen et al 2010). However, it should be noted that the number of events per year does not predict the severity of the next event nor whether the next event will be a potentially life-threatening laryngeal edema (Bowen et al 2010). Therefore, the decision to institute routine prophylaxis should be made on a case-by-case basis. Options for routine prophylaxis include attenuated androgens and C1-INH concentrate.
Attenuated Androgens
Attenuated androgens such as stanozolol, danazol, and oxandrolone are thought to work by increasing hepatic synthesis of C1-INH (Craig 2008). The efficacy of attenuated androgens is dose-dependent and must be adjusted for each patient, with the goal of achieving the lowest effective dose (Bowen et al 2010; Craig 2008). Maximum recommended doses for long-term prophylaxis are 200 mg/day for danazol and 2 mg/day for stanozolol. Dosing can start at higher doses and be tapered to the lowest dose that provides adequate control. It is possible to stop attenuated androgens at intermittent intervals of variable duration and restart when HAE symptoms arise. Danazol is more widely available than stanozolol in Europe. Several studies have shown danazol and stanozolol to be highly effective, reducing the frequency of HAE attacks substantially and rendering many patients free of symptoms (Gelfand et al 1976; Cicardi et al 1991; Bork et al 2008; Sloane et al 2007). However, some studies have suggested that the long-term efficacy of attenuated androgens may wane with time (Füst et al 2011; Varga et al 2009).
Attenuated androgen therapy is associated with numerous dose-dependent side effects, including weight gain, virilization, muscle pains and cramps, headaches, depression, fatigue, GI side effects , menstrual irregularities, and hepatic side effects (Gompels et al 2005; Craig 2008). Such side effects often lead patients to discontinue therapy (Bork et al 2008). Female patients, who are likely to have more frequent and more severe HAE attacks and, therefore, more likely to require prophylaxis, may be particularly sensitive to the virilization side effects of attenuated androgens. Patients should be counseled on the potential side effects of attenuated androgen therapy.
Attenuated androgens should not be used in children and are contraindicated when pregnancy is planned and during pregnancy and lactation. The 2010 International consensus document advises liver function tests be performed every 6 months along with a lipid profile, complete blood count, and urinalysis (Bowen et al 2010).
Pasteurized, Nanofiltered C1-INH Therapy
The nfC1-INH product was recently approved by the EMA for routine prevention in adult and adolescent patients with severe and recurrent attacks who are intolerant to or insufficiently protected by oral treatments, or patients who are inadequately managed with repeated acute treatment. In the United States, the product is approved for routine prophylaxis. For routine prevention of HAE attacks, the recommended dose of nfC1-INH is 1000 U administered every 3 to 4 days. Patients may be provided with 1000 to 2000 U C1-INH concentrate to keep at home which they can take to nearby hospitals for administration if they are not capable of self-administration at home.
Antifibrinolytic Agents
Tranexamic acid and epsilon-aminocaproic acid (EACA) are sometimes used as long-term prophylaxis particularly in prepubertal children, but are less effective than attenuated androgens (Bowen et al 2010). Tranexamic acid is more effective than EACA. The total starting dose of tranexamic acid is 20-50 mg/kg/day, divided as 2 or 3 daily doses (Bowen et al 2010). Gastrointestinal side effects such as dyspepsia are common. A risk of glaucoma has also been reported (Bowen et al 2010).
Considerations for the General Physician
Given the rarity of the condition, the general physician may never see a patient with HAE during his/her career. Nevertheless, an awareness of HAE and its clinical manifestations can help expedite diagnosis and appropriate treatment if a patient does present with recurring episodes of cutaneous swelling without urticaria and episodes of abdominal pain and vomiting. Although HAE patients are best managed by an experienced allergist/immunologist, patients outside major cities and towns or without access to acute treatment may require management by a general physician.
Medications such as ACE inhibitors are contraindicated in patients with HAE. ACE is the primary enzyme that degrades bradykinin; ACE inhibition, therefore, can lead to accumulation of bradykinin, which can worsen HAE attacks (Kaplan and Greaves 2005). Estrogen also induces HAE attacks; estrogen-containing oral contraceptives and hormone replacement therapy often lead to worsening of attacks. Women requiring oral contraceptives should be prescribed progestogen-only formulations, and estrogen-containing hormone replacement therapy should be avoided in postmenopausal women (Bowen et al 2010).
HAE patients are likely to receive plasma-derived products, including C1-INH concentrate. Physicians should ensure that such patients have been vaccinated against bloodborne pathogens such as hepatitis B (Bowen et al 2010).
It should be noted that the designated units of C1 esterase in plasma-derived and recombinant C1-INH preparations are not comparable. These preparations are also of different vial sizes. The dose required for treatment is calculated on the basis of units per kilogram of body weight. Although in practice it should be possible to use these products by rounding up the dose to the nearest whole vial, clinicians should consult the labeling of the products to determine appropriate doses.
Psychological stress is also known to be a trigger of HAE attacks. Patients may benefit from counselling to manage and reduce psychological stress.
In 75% of cases, HAE is inherited in an autosomal dominant pattern. Family members of patients should undergo testing even if they have not yet exhibited symptoms. In a survey of 457 HAE patients in the United States, only about half of patients’ immediate family members and about one quarter of extended family members had been tested for HAE (Lunn et al 2010).
Finally, general physicians should be aware that even minor trauma (eg, muscle pull) and minor manipulations (eg, tooth extraction) can trigger HAE attacks in some patients. Patients should be made aware of the elevated risk of HAE attacks during minor and major medical and surgical procedures, and physicians should ensure that short-term prophylaxis against HAE attacks is instituted if deemed necessary. HAE patients may also be at greater risk of developing autoimmune disorders and require long-term follow-up.
Summary
HAE is a rare but potentially life-threatening inherited disorder that has a significant impact on the quality of life and productivity of affected individuals. The highly variable clinical presentation of HAE can lead to significant delays in diagnosis, leaving many patients untreated. Diagnosis may be made based on clinical and patient factors (ie, repeated episodes of cutaneous swelling and of abdominal pain and vomiting, onset of symptoms at puberty or young adulthood, family history of such episodes) and confirmed with laboratory assessments of complement proteins. Treatment of HAE is ideally managed by an experienced allergist/immunologist, but general physicians may need to manage patients who do not have access to specialist care. In addition, general physicians need to be aware of the non–disease-specific needs of HAE patients, including medications to be avoided, possible need for help/treatment to cope with anxiety/depression, need for prompt management of certain infections, and precautions to consider before medical, dental, or surgical procedures. Increased awareness of HAE among general physicians can help identify patients who may have HAE and expedite their diagnosis and appropriate treatment.
Acknowledgments
The author wishes to thank Euro RSCG Life Catapult for editorial support in the preparation of the manuscript, which was funded by ViroPharma Incorporated.
References:
Agostini A, Aygoren-Pursun E, Binkley K, et al. (2004) Hereditary and acquired angioedema: Problems and progress: Proceedings of the third C1 esterase inhibitor deficiency workshop and beyond. Journal of Allergy and Clinical Immunology. 114, S51-S131.
Binkley K. (2010) Factor XII mutations, estrogen-dependent inherited angioedema and related conditions. Allergy, Asthma and Clinical Immunology. 6, 16.
Bork K, Siedlecki K, Bosch S, Schopf RE, Kreuz W. (2000a) Asphyxiation by laryngeal edema in patients with hereditary angioedema. Mayo Clinic Proceedings. 75, 349-354.
Bork K, Barnstedt S, Koch P, Traupe H. (2000b) Hereditary angioedema with normal C1-inhibitor activity in women. Lancet. 356, 213-217.
Bork K, Meng G, Staubach P, Hardt J. (2005). Treatment with C1 inhibitor concentrate in abdominal pain attacks of patients with hereditary angioedema. Transfusion. 45,1774-1784.
Bork K, Meng G, Staubach P, Hardt J. (2006a). Hereditary angioedema: New findings concerning symptoms, affected organs, and course. American Journal of Medicine. 119, 267-274.
Bork K, Staubach P, Eckardt AJ, Hardt J. (2006b). Symptoms, course, and complications of abdominal attacks in hereditary angioedema due to C1 inhibitor deficiency. American Journal of Gastroenterology.101, 619-627.
Bork K, Gül D, Hardt J, Dewald G. (2007) Hereditary angioedema with normal C1 inhibitor: clinical symptoms and course. American Journal of Medicine. 120, 987-992. Bork K, Bygum A, Hardt J. (2008) Benefits and risks of danazol in hereditary angioedema: a long-term survey of 118 patients. Annals of Allergy, Asthma and Immunology. 100, 153-161. Bork K. (2010) Diagnosis and treatment of hereditary angioedema with normal C1 inhibitor. Allergy, Asthma, and Clinical Immunology. 6,15.
Bouillet L, Boccon-Gibod I, Ponard D, et al. (2009) Bradykinin receptor 2 antagonist (icatibant) for hereditary angioedema type III attacks. Annals of Allergy, Asthma & Immunology. 103, 448.
Bowen T, Cicardi M, Bork K, et al. (2008) Hereditary Angioedema: a current state-of the-art review, VII: Canadian-Hungarian 2007 International Consensus Algorithm for the Diagnosis, Therapy and Management of Hereditary Angioedema. Annals of Allergy, Asthma and Immunology. 100(Suppl 2):S30-S40.
Bowen T (2011). Hereditary angioedema: beyond international consensus—circa December 2010—The Canadian Society of Allergy and Clinical Immunology Dr. David McCourtie Lecture. Allergy, Asthma and Clinical Immunology. 7, 1.
Bowen T, Cicardi M, Farkas H, et al. (2010) 2010 International consensus algorithm for the diagnosis, therapy and management of hereditary angioedema. Allergy, Asthma, and Clinical Immunology. 6,24.
Brickman CM, Tsokos GC, Balow JE, et al. Immunoregulatory disorders associated with hereditary angioedema I: clinical manifestations of autoimmune disease. (1986) Journal of Allergy and Clinical Immunology. 77, 749-757.
Cicardi M, Zanichelli A. (2010) Acquired angioedema. Allergy, Asthma and Clinical Immunology. 6, 14.
Cicardi M, Bergamaschini L, Cugno M, Hack E, Agostoni G, Agostoni A. (1991) Long-term treatment of hereditary angioedema with attenuated androgens: a survey of a 13-year experience. Journal of Allergy and Clinical Immunology. 87, 768-773. Cicardi M, Banerji A, Bracho F, et al. (2010) Icatibant, a new bradykinin-receptor antagonist, in hereditary angioedema. New England Journal of Medicine. 363, 532-541. Cicardi M, Bork K, Caballero T, et al. (2012). Evidence-based recommendations for the therapeutic management of angioedema owing to hereditary C1 inhibitor deficiency: consensus report of an International Working Group. Allergy. 67, 147-157. Craig TJ. (2008) Appraisal of danazol prophylaxis for hereditary angioedema. Allergy Asthma Proceedings. 29, 225-231.Donaldson VH, Evans RR. (1963) A biochemical abnormality in hereditary angioneurotic edema: absence of serum inhibitor of C1-esterase. American Journal of Medicine. 35, 37-44.
Füst G, Farkas H, Csuka D, Varga L, Bork K. (2011) Long-term efficacy of danazol treatment in hereditary angioedema. European Journal of Clinical Investigation. 41, 256-262. Gelfand JA, Sherins RJ, Alling DW, Frank MM. (1976) Treatment of hereditary angioedema with danazol. Reversal of clinical and biochemical abnormalities. New England Journal of Medicine. 295, 1444-1448.
Gompels MM, Lock RJ, Abinum M, et al. (2005) C1 inhibitor deficiency: consensus document. Clinical and Experimental Immunology. 139, 379-394.
Gösswein T, Kocot A, Emmert G, et al.(2008) Mutational spectrum of the C1INH (SERPING1) gene in patients with hereditary angioedema. Cytogenetics and Genome Research. 121, 181-188.Kaplan AP, Greaves MW. (2005) Angioedema. Journal of the American Academy of Dermatology. 53, 373-388.
Kaplan AP, Joseph K. (2010) The bradykinin-forming cascade and its role in hereditary angioedema. Annals of Allergy, Asthma, and Immunology. 104, 193-204.
Lumry WR, Castaldo AJ, Vernon MK, Blaustein MB, Wilson DA, Horn PT. (2010) The humanistic burden of hereditary angioedema: Impact on health-related quality of life, productivity, and depression. Allergy and Asthma Proceedings. 31, 407-414.
Lumry WR, Busse P, Baker J, et al. (2011) Pre-procedure administration of C1 esterase inhibitor (human) (Cinryze) for the prevention of hereditary angioedema (HAE) attacks after medical, dental, or surgical procedures. Presented at the 2011 annual meeting of the American Academy of Allergy, Asthma & Immunology. San Francisco, California, March 18-22, 2011; Abstract 903.
Lunn ML, Santos CB, Craig TJ. (2010) Is there a need for clinical guidelines in the United States for the diagnosis of hereditary angioedema and the screening of family members of affected patients. Annals of Allergy, Asthma and Immunology. 104, 211-214.
Martin L, Raison-Peyron N, Nothen MM, Cichon S, Drouet C. (2007) Hereditary angioedema with normal C1 inhibitor gene in a family with affected women and men is associated with the p.Thr328Lys mutation in F12 gene. Journal of Allergy and Clinical Immunology. 120, 975-977.
Nzeako U, Frigas E, Tremaine WJ. (2001) Hereditary angioedema: A broad review for clinicians. Archives of Internal Medicine. 161, 2417-2429.
Osler W. (1888) Hereditary angioneurotic edema. American Journal of Medical Science. 95, 362-367.
Pickering PJ, Good RA, Kelly JR. (1969) Replacement therapy in hereditary angioedema: successful treatment of two patients with fresh frozen plasma. Lancet. 293, 326-330.
Prematta M, Gibbs JG, Pratt EL, Stoughton TR, Craig TJ. (2007) Fresh frozen plasma for the treatment of hereditary angioedema. Annals of Allergy, Asthma, and Immunology. 98,383-388.Quincke H. (1882) Uber akutes umschriebens Hautoderm. Monatschrift Praktische Dermatologie. 1, 129-131.
Reshef A, Leibovich I, Goren A. (2008) Hereditary angioedema: new hopes for an orphan disease. Israeli Medical Association Journal. 10, 850-855.Rusicke E, Martinez-Saguer I, Aygören-Pürsün, et al. (2010) Experiences of short-term prophylaxis in adult and pediatric patients with hereditary angioedema prior to surgical procedures. Presented at the 29th Congress of the European Academy of Allergy and Clinical Immunology. London, United Kingdom, June 5-9, 2010. Abstract 1183.
Sloane DE, Lee CW, Sheffer AL. (2007) Hereditary angioedema: Safety of long term stanozolol therapy. Journal of Allergy and Clinical Immunology. 120, 654-658.
Varga L, Csuka D, Füst G, Farkas H. (2009) The effect of long-term danazol therapy on the serum level of complement proteins and attack frequency in hereditary angioedema. Molecular Immunology. 46, 2871.
Zingale LC, Castelli R, Zanichelli A, Cicardi M. (2006) Acquired deficiency of the inhibitor of the first complement component. Presentation, diagnosis, course and conventional management. Immunology and Allergy Clinics of North America. 26, 669-90.
Zuraw BL, Herschbach J. (2000) Detection of C1 inhibitor mutations in patients with hereditary angioedema. Journal of Allergy and Clinical Immunology. 105, 541-546. Zuraw B, Cicardi M, Levy RJ, et al. (2010). Recombinant human C1-inhibitor for the treatment of acute angioedema attacks in patients with hereditary angioedema. J Allergy Clin Immunol 2010;126:821-827.
*Cetor is a plasma-derived pasteurized human C1 inhibitor esterase manufactured by Sanquin, the Netherlands, which holds the Market Authorisation, and is available in the Netherlands, and in Belgium, Finland, France, and Luxembourg by mutual recognition procedure.
Jimmy H.C. Gooi, MD, FRCP
Consultant Clinical Immunologist, St James’s University Hospital, Leeds, United Kingdom & Beaumont Hospital, Dublin, Ireland
Address correspondence to:
Department of Immunology
Beaumont Hospital
PO Box 1297, Dublin 9
Ireland
First Published February 2012
Copyright Priory Lodge Education Limited 2012 -
All pages copyright ©Priory Lodge Education Ltd 1994-


