Chronic Bronchitis only really became recognised as a distinct disease rather than a set of symptoms in the late 1950's. The great British Smogs of the 1950's precipitated the deaths of many patients from respiratory failure, and on the continent, chronic bronchitis was referred to as the English Disease. There can be little doubt now that the most important risk factor in the development of COPD is cigarette smoking. The effects of cigarette smoke on the lung are manifold. Cigarette smoke has been found to attract inflammatory cells into the lungs and stimulates the release of of the proteolytic enzyme elastase from these cells. Elastase breaks down elastin, a normal structural component of lung tissue, but normally, the lung is protected from the destructive effect of elastase by an inhibitor, alpha-1 antitrypsin (AAT). However, cigarette smoke attracts more cells and stimulates the release of more elastase than can be inhibited by the circulating levels of AAT. In addition, cigarette smoke itself may inactivate AAT therefore swinging the balance in favour of more lung destruction by elastase. The development of COPD , and in particular emphysema, is thought to be due to the imbalance between the destructive elastase and protective AAT.
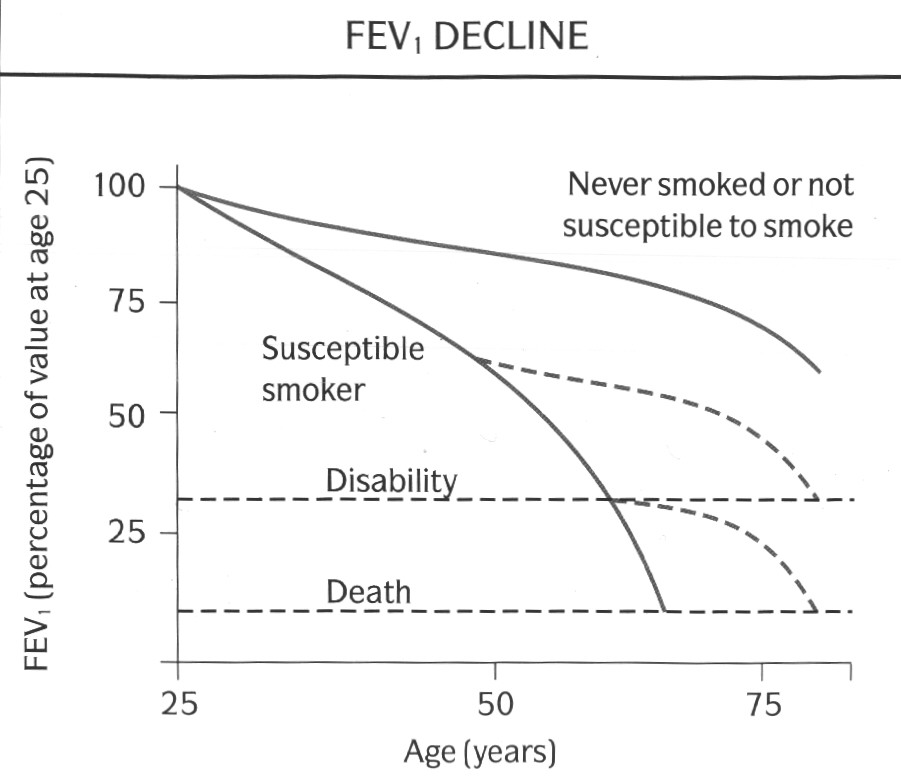
Not all people who smoke, however, develop COPD; and not all patients with COPD are smokers or have smoked in the past. There seems to be a varying susceptibility to lung damage due to cigarette smoke within the general population. Only a proportion of smokers (maybe only 10-15%) show a rate of decline of lung function over the years that is fast enough to result in the severe impairment that is typical of patients who present with breathlessness due to COPD. Susceptible subjects have an accelerated rate of decline of lung function (50-90ml of FEV1/yr compared with 20-30ml of FEV1/yr after the age of 30 in non-smokers). Subjects with COPD who stop smoking slow down the progression of disease and may return to normal levels of FEV1 decline. Unfortunately, they do not improve after they stop smoking (fig 3). By the time subjects are symptomatic with breathlessness, they will have already have severe impairment of lung function, and stopping smoking at this stage may extend their life expectancy but may not improve their symptoms.
(Fig. 3: graph of survival from Fletcher C, Peto R, Br Med J, 1:1645-1648, 1977, Reproduced with permission)
One possibility to account for these differences is that there is a genetically determined predisposition to develop allergy and bronchial hyperresponsiveness, the "Dutch Hypothesis". According to this, asthma, emphysema and chronic bronchitis are different manifestations of a single disease process. Whether an individual develops asthma, bronchitis or emphysema is a result of genetic and environmental factors that are modulated by age and gender. An alternative school of thought is the "Two-type Hypothesis", which includes a Dutch-type limb termed "Chronic Asthmatic Bronchitis" or "Overlap Syndrome", and a more insidious form which leads to "Chronic Obstructive Bronchitis and Emphysema". Both schools of thought, however, emphasise the inter-relationship between bronchial hyperreactivity (atopy), infection and smoking (fig.4). Focus has recently been placed in trying to identify the population most at risk of developing COPD.
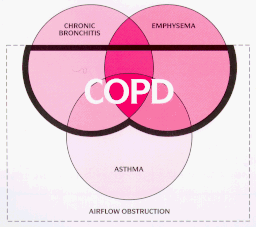
Fig. 4 : Venn
diagram of overlap between asthma, chronic bronchitis and
emphysema
(the diagram is not proportional)
Another well established risk factor is deficiency of the protective protease inhibitor, Alpha-1 Antitrypsin (AAT). This is an inherited autosomal recessive (designated PiZZ) disorder which is fairly rare in the general gene pool. The estimated prevalence of the heterozygote phenotype (PiMZ) is about 2-3% of the western population. The incidence of homozygous births is in the region of 1 in 3000 live births. As such, AAT deficiency account for probably less than 5% of all cases of COPD.
Low levels of AAT allow the uninhibited action of elastase on the lung parenchyma giving rise to destruction of the alveoli and the eventual development of emphysema rather than chronic bronchitis. The pattern of emphysema in AAT deficiency differs slightly from that of smoking induced pure emphysema in that AAT deficiency produces panlobular emphysema affecting predominantly the lower lung fields, and smoking induced emphysema is usually centrilobular affecting the upper lung fields initially. Not all people with PiZZ have very low levels of AAT. Only serum levels below 35% normal are at risk of developing emphysema. Subjects who are PiMZ may also have reduced levels but not as low as PiZZ, and are not thought to be at more risk from developing emphysema than PiMM subjects. Subjects who are PiZZ may not progress to have emphysema, but PiZZ subjects who smoke have a greatly increased risk of developing emphysema, especially at an early age (Fig 5).
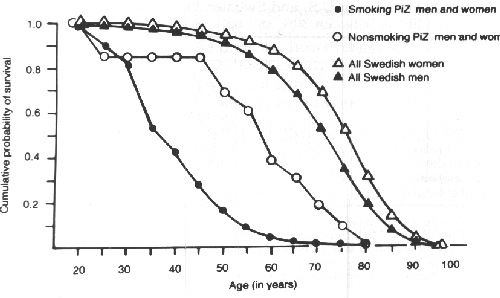
(Fig. 5: Graph of
cumulative survival from Larsson C. Acta Med. Scand. 204:345-351,
1978, reproduced with permission)
The role of outdoor air pollution in the evolution of COPD is still controversial. Respiratory deaths in the UK reached a peak during the great smogs of the 1950's. Following the passing of the Clean Air Acts of 1956 and 1968 which established "smokeless zones" in populated areas and allowing only the use of smokeless fuels, the quality of British air has improved. The people that died during the smogs were people at the greatest risk, i.e. the elderly and infirmed, and those with chronic respiratory and cardiac problems. The question of whether atmospheric pollution itself can cause or contribute to the development of COPD is still uncertain.
Outdoor air pollution is very heterogeneous and is different in different areas. It is mainly comprised of particulates and gases with some background radioactivity. The particulates mainly originate from the incomplete combustion of solid fuels and diesel, ash and fine dusts. The main gaseous components are the various oxides of sulphur, nitrogen and carbon, again from the combustion of fossil fuels; hydrocarbons and ozone. Studies from the UK have shown a relationship between levels of atmospheric pollution and respiratory problems (particularly cough and sputum production) in both adults and children, and similar studies from the USA have confirmed these findings. Some studies have reported lower levels of lung function in adults living in highly polluted areas and this seems to related to pollution by acidic gases and particulates. As with the problem of smoking, there will be individuals who are more susceptible to the effects of atmospheric pollution than others.
Any occupation in which the local environment is polluted with the aforementioned gases and particulates increases the risk of developing of COPD. In addition, there is evidence that cadmium and silica also increase the risk of COPD. This is especially true if the subject smokes. Occupations at risk include coal miners, construction workers who handle cement, metal workers, grain handlers, cotton workers and workers in paper mills. However, the effect of smoking far outweighs any influences from the work environment.
Most of the tobacco smoke in a room it that which is coming form the burning end of the cigarette rather than the smoke exhaled from the smoker's lungs. This smoke (called sidestream smoke) is actually higher in concentration of toxic substances than exhaled smoke (mainstream smoke). However, it has been very difficult to judge how much smoke is passively inhaled and what effects this passively inhaled smoke has on the lungs. Studies on passive smoking are plagued by methodological difficulties. Studies in which questionnaires are used to assess the degree of passive exposure to cigarette smoke are prone to bias. Recently, it has been possible to assess the degree of exposure by measuring levels of the nicotine metabolite, cotinine, either in the blood, saliva or urine. Most of the studies using these techniques have been cross sectional ones on children from smoking or non-smoking families. The evidence suggests that respiratory infections and respiratory symptoms are more common in children in households where one or both parents smoke. Also, there is a small but significant difference in the prevalence of respiratory symptoms and lung function in adults and children who are regularly exposed to passive smoking. Whether these differences are clinically significant is yet to be resolved.
The role of viral infections of upper and lower respiratory tract in the pathogenesis of COPD remains to be clarified. Viral infections in the lung enhance inflammation and predispose to bronchial hyperreactivity. There is increasing evidence between early childhood infections and increase in respiratory symptoms and lower lung function in adulthood. The viruses that have been implicated are adenovirus and respiratory syncytial virus. Once COPD is established, repeated infective exacerbations of airflow obstruction, either viral or bacterial, may speed up the decline in lung function.
Chinese and Afro-Caribbean races seem to have a reduced susceptibility to developing COPD. It is frequently stated that COPD is more prevalent in men. However, when smoking and occupational exposure is taken into account, the relative risk of developing COPD is not significantly higher in men than women. With smoking on the increase in women, it is possible that women may catch up with men in terms of absolute numbers. The beneficial effects of stopping smoking on the rate of lung function decline may greater for women than men.
In studies conducted in the UK in the 1950's and 1960's, there
is a clear social class gradient for COPD with it being more
prevalent in the lower socioeconomic strata. This may be related
to poorer housing and nutrition and use of fossil fuels for
heating without adequate ventilation. Also, there is a higher
prevalence of smoking in the lower socioeconomic strata, and they
are more likely to be employed in jobs where they may be a risk
from occupational exposure. However, this socioeconomic gradient
fro COPD is now becoming more smoothed out as standards of living
improve.